1写在前面
之前我们介绍的基于发生率或者均值进行样本量计算的方法,但都是在组间进行计算。🤔
有的时候我们需要获取组内变化,进行样本量计算。🤫
常见的就是配对样本,比如相同受试者进行多个时间点的观察,如下图:👇
本期我们就介绍一下如何估算配对样本的样本量吧。🥰
2用到的包
rm(list = ls())
library(pwr)
library(tidyverse)
3研究假设
还是假设我们正在进行一项前瞻性研究,测量一组开始节食患者的体重变化。🧐
我们先提出研究假设,
和
:👇
: 该组患者 基线和节食3周后体重的平均变化没有差异。: 该组患者 基线和节食3周后体重的平均变化存在差异。
4计算样本量
现在我们假设需要招募足够多的受试者,以检测节食开始3周后体重减轻了5磅。⤵️
假设基线平均体重为130磅,标准差为11,节食3周后,预期平均体重为125磅,标准差为12。🤪
4.1 计算Cohen’s d
mu_x <- 130 ### Baseline体重均值
mu_y <- 125 ### diet后体重均值
sd_x <- 11 ### Basline体重SD
sd_y <- 12 ### diet后体重SD
rho <- 0.5 ### diet前后相关性
sd_z <- sqrt(sd_x^2 + sd_y^2 - 2*rho*sd_x*sd_y)
d_z <- abs(mu_x - mu_y) / sd_z
d_z

Note! 顺便说一下,有时候我们不知道这个相关性rho是多少,可以默认设置成0.5。🤓
4.2 pwr计算样本量
现在,我们可以利用pwr包计算节食前后平均体重变化差(5磅)所需的样本量,具有 80%的power和0.05的显著性。🥳
这里我们可以看到需要44例受试者,进行2次体重检测。
n.paired <- pwr.t.test(d = d_z, power = 0.80, sig.level = 0.05, type = "paired")
n.paired

5Power Analysis
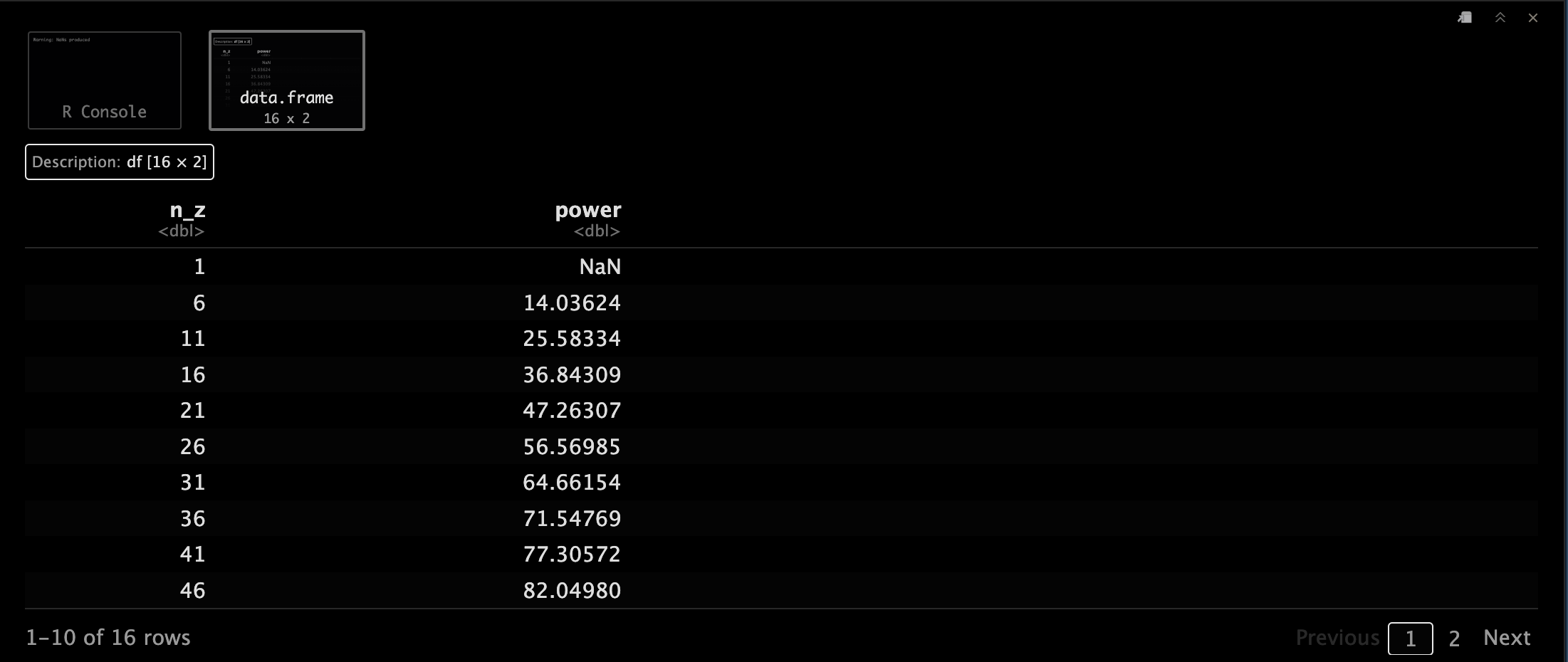
接着是效力分析(Power Analysis),配对t检验的样本量变化对power的影响吧。🤜
与之前的示例一样,随着我们增加样本量,估算的不确定性也随之减小。🧐
通过减少这种不确定性,我们在估算中更好地避免了II类错误。😙
n_z <- seq(1, 80, 5)
n_z.change <- pwr.t.test(d = d_z, n = n_z, sig.level = 0.05, type = "paired")
n_z.change.df <- data.frame(n_z, power = n_z.change$power * 100)
n_z.change.df
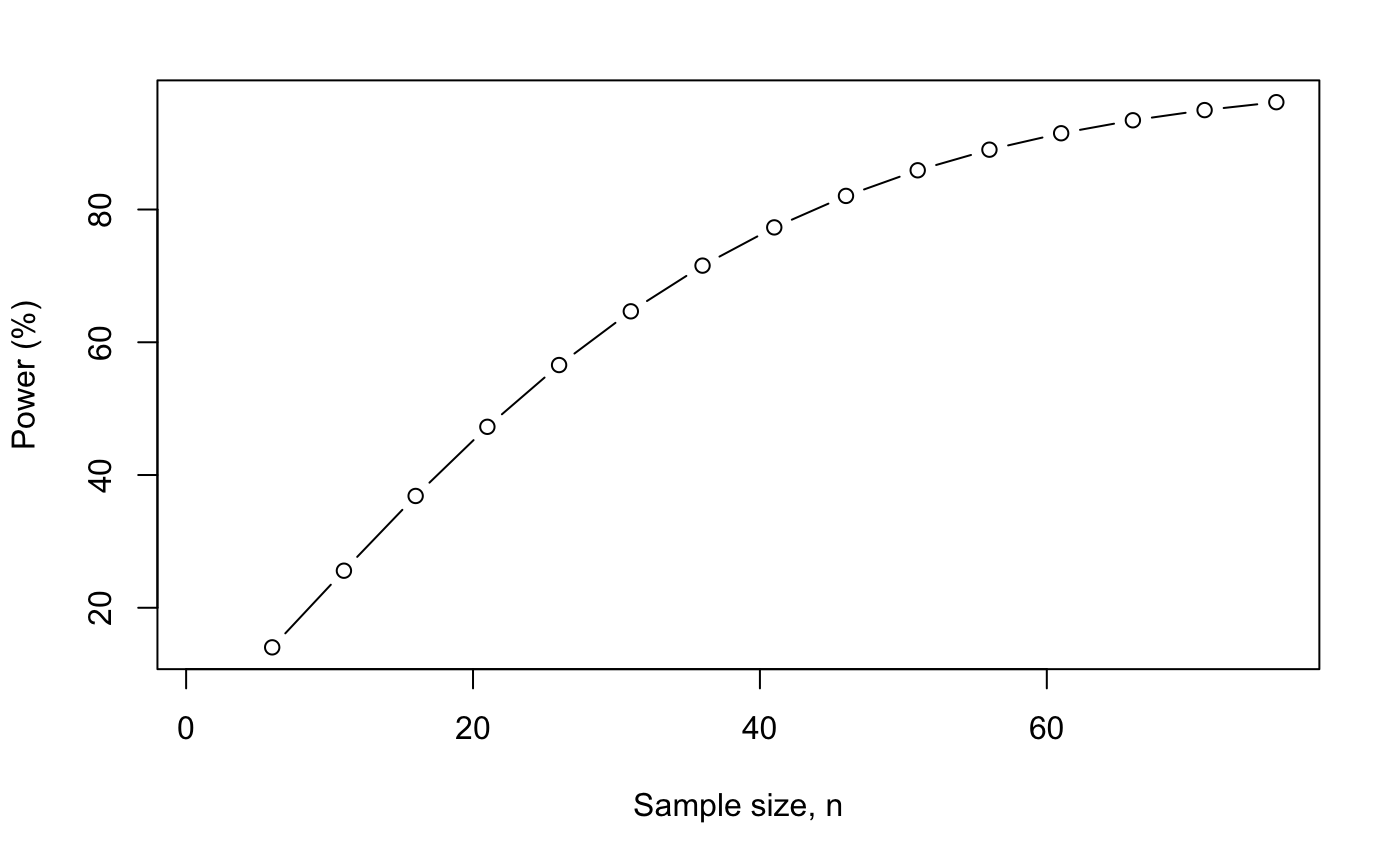
plot(n_z.change.df$n,
n_z.change.df$power,
type = "b",
xlab = "Sample size, n",
ylab = "Power (%)")
6改变一下
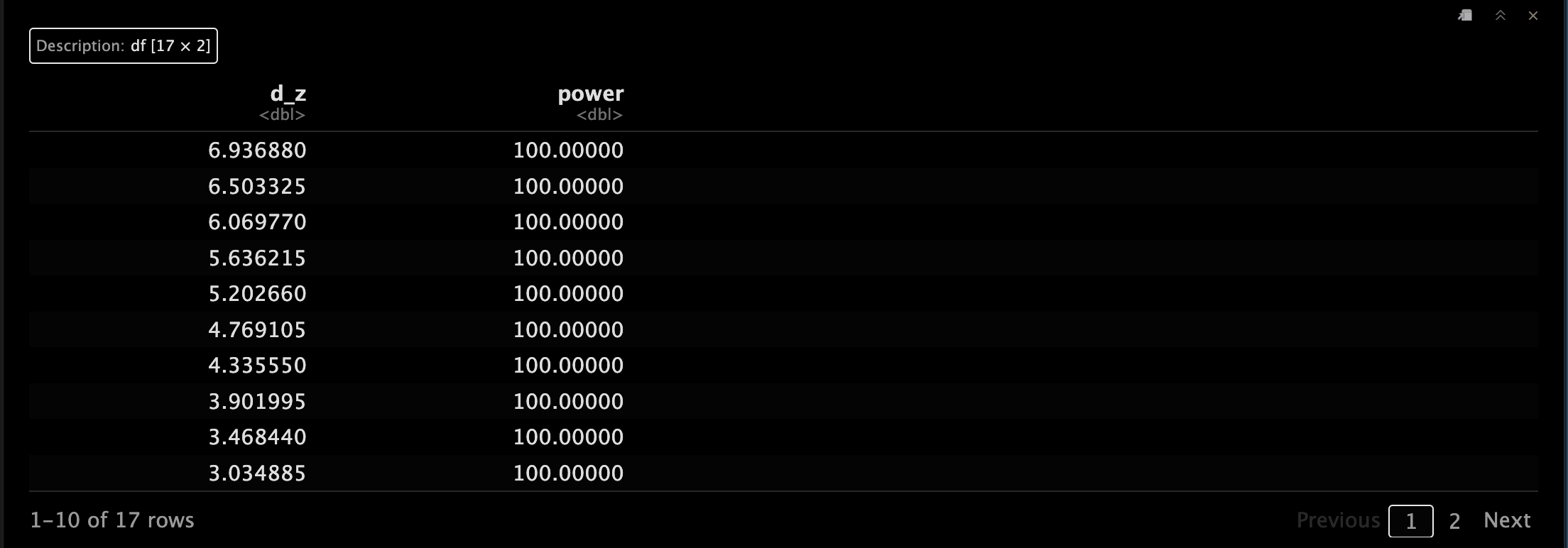
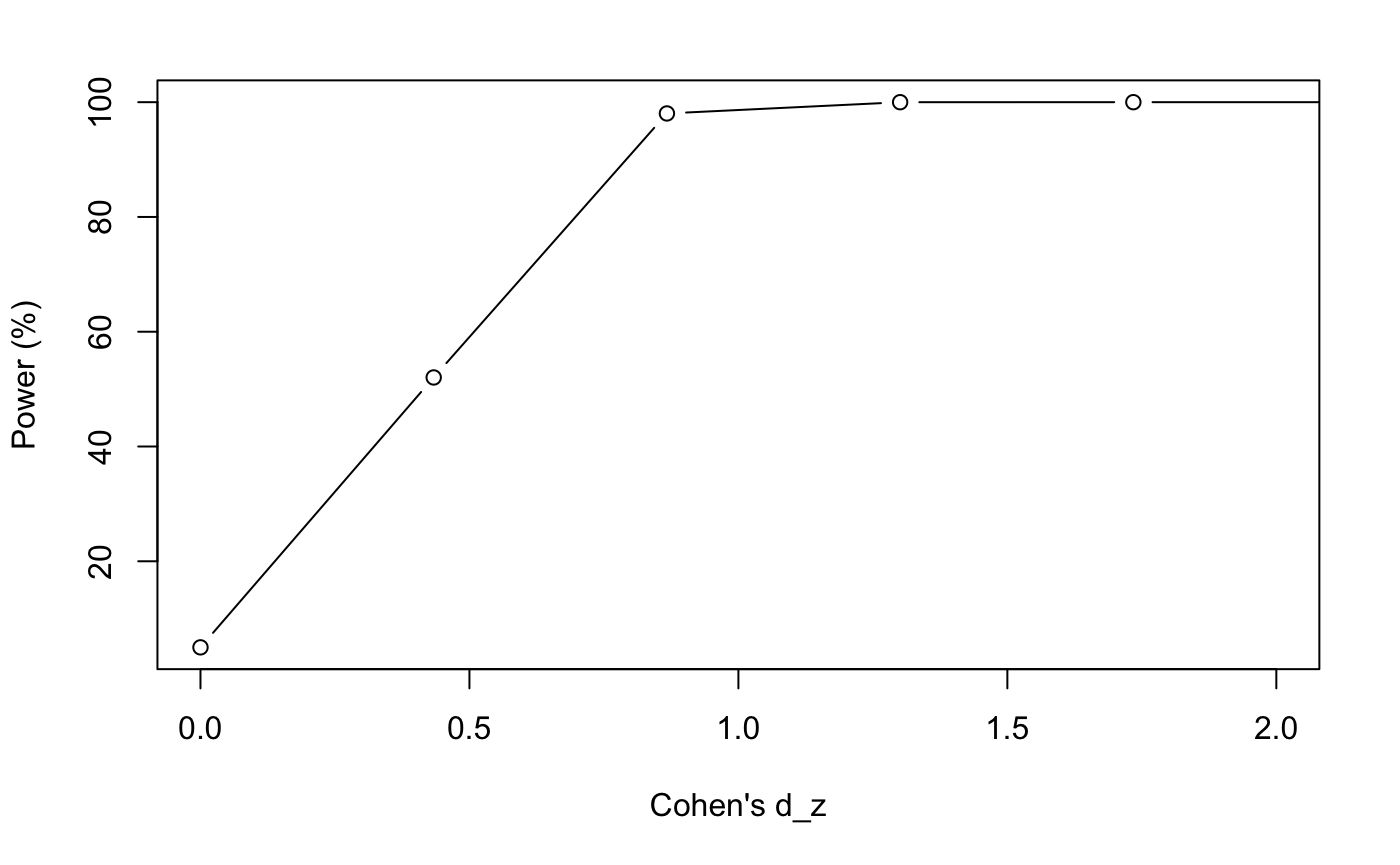
让我们看看当我们改变effect size时power如何变化。😗
接着我们改一下患者节食导致的平均体重变化,看看减少到100磅时power如何变化。😂
这里我们从50磅开始,逐渐增加到130磅,间隔5磅。😉
mu_y <- seq(50, 130, 5)
d_z <- abs(mu_x - mu_y) / sd_z
n_z.change <- pwr.t.test(d = d_z, n = 44, sig.level = 0.05)
n_z.change.df <- data.frame(d_z, power = n_z.change$power * 100)
n_z.change.df
plot(n_z.change.df$d_z,
n_z.change.df$power,
type = "b",
xlab = "Cohen's d_z",
ylab = "Power (%)",
xlim = c(0, 2))
7不等比样本量的Power Analysis
有时候我们做的研究,两组并不是等比的,这个时候就非常需要做Power Analysis了。😘
假设我们现在准备做一个回顾性研究(Retrospective study),其中患者分别接受了Treatment A和Treatment B。🤨
Treatment A有130名患者 (nA = 130),Treatment B有120名患者 (nB = 120)。😜
在Treatment A中,HbA1c的平均变化为1.5%,标准差为1.25%。🤫
在Treatment B中,HbA1c的平均变化为1.4%,标准差为1.01%。🫢
7.1 计算并合标准偏差
首先我们计算一下并合标准偏差(pooled standard deviation, σpooled)。😂
sd1 <- 1.25
sd2 <- 1.01
sd_pooled <- sqrt((sd1^2 +sd2^2) / 2)
sd_pooled

7.2 计算Cohen’s d
mu1 <- 1.5
mu2 <- 1.4
d <- (mu1 - mu2) / sd_pooled
d

7.3 Power Analysis
这个时候我们只有11%的power来可以发现HbA1c与基线相比变化0.10%或更大的差异。🤠
也就是说在nA = 130和nB = 120以及显着性水平为0.05的情况下,我们是发现不了HbA1c变化与基线的0.10%或更大的差异。🫠
这个时候我们如果还要做这个研究,就很可能会出现II类错误。😅
解决这个问题的唯一方法是招募更多患者或通过放宽纳入标准来扩大样本量。🤪
n1 <- 130
n2 <- 120
power.diff_n <- pwr.t2n.test(d = d, n1 = n1, n2 = n2, sig.level = 0.05)
power.diff_n

8补充一下
8.1 其他功能
这里我们补充pwr包里的其他功能,不过可能不是特别常用,主要还是看你的研究设计。🤩
| Function | Power Calculation For |
|---|---|
| pwr.2p.test | two proportions equal n |
| pwr.2p2n.test | two proportions unequal n |
| pwr.anova.test | balanced one way anova |
| pwr.chisq.test | chi square test |
| pwr.f2.test | general linear model |
| pwr.p.test | proportion one sample |
| pwr.r.test | correlation |
| pwr.t.test | t-tests (one sample, 2 samples, paired) |
| pwr.r.test | t-test (two samples with unequal n) |
8.2 Effect size
确定Effect size有时候真的特别难,这里提供一些经验性的参考吧。🥸
| Effect size | Cohen’s w |
|---|---|
| Small | 0.10 |
| Medium | 0.30 |
| Large | 0.50 |

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......
本文由 mdnice 多平台发布