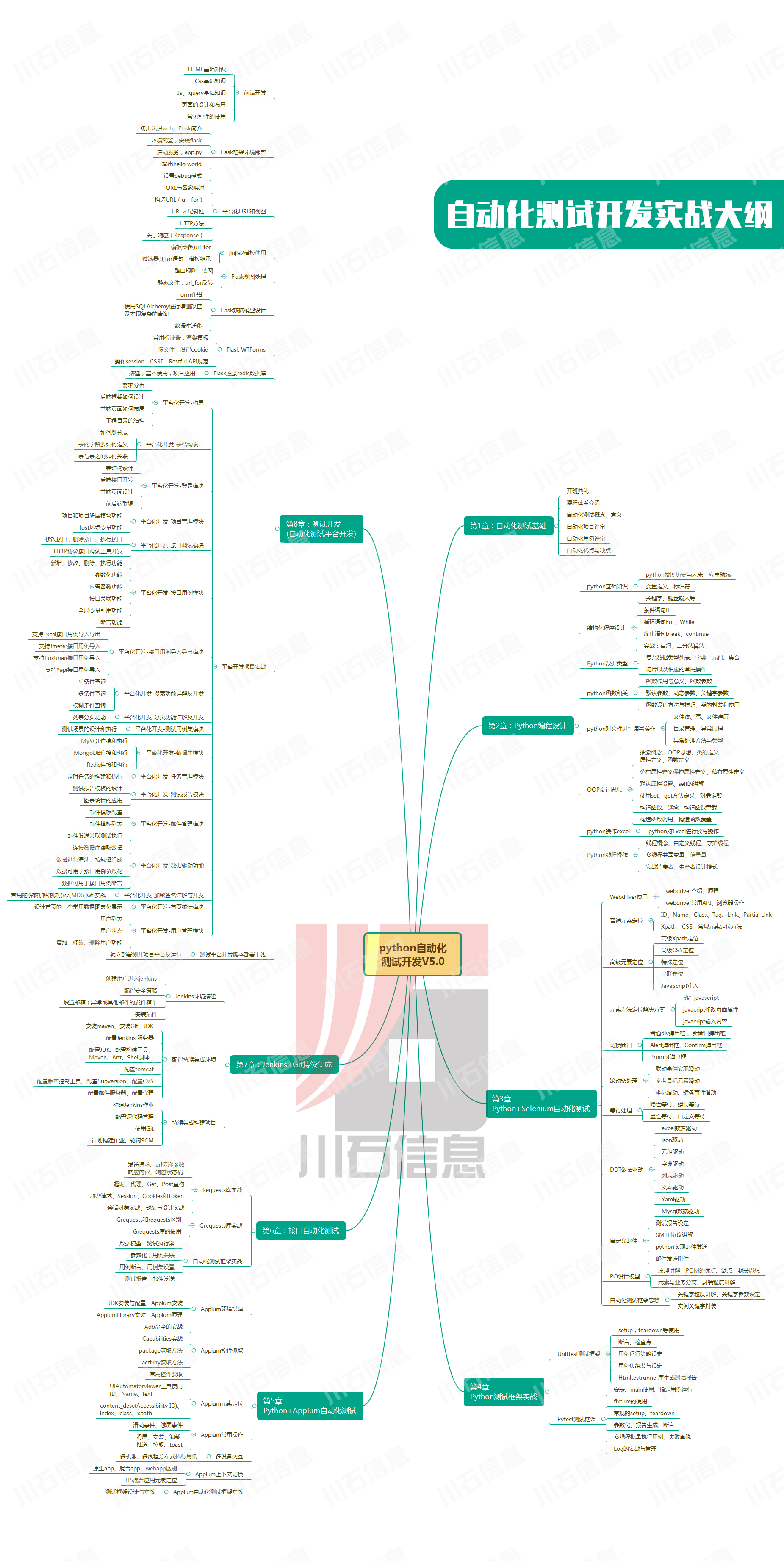
ldsc跨物种计算
news2026/4/29 20:51:08
library(Seurat) library(dplyr) library(data.table) library(GenomicFeatures) library(parallel) library(clusterProfiler) #library(tidyverse) library(patchwork) #####分细胞类型进行ldsc计算 ###1.准备猴与人类同源gene转换 ## 以人和食蟹猴为例此处需要联网操作本地操作 library(biomaRt) library(dplyr) library(stringr)数据处理HIP - readRDS(/data/users/laizilin/laizilin_c11721fec6dd4fa0833515e5103c7fb7/online/data/cortex/A1/Cortex.A1.5data.combined.new0302.rds) # 将默认 Assay 切换为 RNA DefaultAssay(HIP) - RNA # 运行后可以直接输入对象名查看 Active assay 是否已经变成了 RNA HIP # 如果 species 是 BGI_Human 或 Public_Human就改成 Human否则保持原样 HIP$species.V1 - ifelse(HIP$species %in% c(BGI_Human, Public_Human), Human, HIP$species) # 验证一下结果 table(HIP$celltype.V1, HIP$species.V1) # 将 空格、、- 统一替换为点号 HIP$celltype.V1 - gsub([ -], ., HIP$celltype.V1) # 把可能出现的连续两个点比如原来是 会变成 ...压缩成一个点更美观 HIP$celltype.V1 - gsub(\\., ., HIP$celltype.V1) # 检查一下结果 table(HIP$celltype.V1, HIP$species.V1)计算deg每个物种分开处理,第二个是取前2000deg做法library(Seurat) library(dplyr) library(future) # 加载并行计算包 # 多线程配置区 # 1. 设置使用的 CPU 核心数 (建议根据你的服务器配置设置比如 4、8、16) # 注意核心数越多消耗的内存 (RAM) 呈倍数增长量力而行。 plan(multisession, workers 4) # 2. 解除内存限制 (极其重要) # Seurat 并行计算时需要把数据复制给每个核心。如果数据大默认的 500MB 限制会报错。 # 这里设置为 50GB (50 * 1024^3)你可以根据服务器内存大小调整如 100GB options(future.globals.maxSize 50 * 1024^3) # # 参数配置区 col_celltype - celltype.V1 col_species - species.V1 # # 1. 强制使用 RNA assay DefaultAssay(HIP) - RNA # 2. 动态生成带有物种后缀的组合列并设为 Idents HIP$celltype_species - paste0(HIP[[col_celltype]][,1], _, HIP[[col_species]][,1]) Idents(HIP) - celltype_species # 3. 获取物种列表准备空列表装结果 species_list - unique(HIP[[col_species]][,1]) all_markers_list - list() # 4. 开始循环找 Marker (现在它会自动多线程狂飙了) for (sp in species_list) { cat(\n\n) cat( 正在多线程寻找物种的 Marker:, sp, ...\n) # 切出当前物种的子集 sub_obj - HIP[, HIP[[col_species]][,1] sp] # 切掉 Idents 里的 _物种名 尾巴 Idents(sub_obj) - gsub(pattern paste0(_, sp, $), replacement , x Idents(sub_obj)) # 跑 FindAllMarkers底层会自动调用刚才分配的 8 个核心 markers - FindAllMarkers( sub_obj, only.pos TRUE, min.pct 0.25, logfc.threshold 0.5 ) # 找到了就贴上后缀存起来 if (nrow(markers) 0) { markers - markers %% mutate(cluster paste0(cluster, _, sp)) all_markers_list[[sp]] - markers } } # 5. 合并导出 final_deg_df - do.call(rbind, all_markers_list) setwd(/data/work/ldsc_input/) write.csv(final_deg_df, a1_all_celltype_markers2human.csv, row.names FALSE) cat(\n 恭喜多线程提速完成所有物种的 DEG 提取完毕\n)library(Seurat) library(dplyr) # 确保单线程 # 强制设置为顺序执行不使用并行 # library(future) # 如果没加载可以不加 # plan(sequential) # # 1. 基础配置 col_celltype - celltype.V1 col_species - species.V1 DefaultAssay(HIP) - RNA # 假设你已经手动跑过 NormalizeData(HIP) HIP$celltype_species - paste0(HIP[[col_celltype]][,1], _, HIP[[col_species]][,1]) Idents(HIP) - celltype_species species_list - unique(HIP[[col_species]][,1]) all_markers_list - list() # 2. 单线程循环处理 for (sp in species_list) { cat(\n , rep(, 40), \n, sep ) cat( 当前正在处理物种 [单线程]:, sp, \n) # 提取物种对应的子集 sub_obj - HIP[, HIP[[col_species]][,1] sp] Idents(sub_obj) - gsub(pattern paste0(_, sp, $), replacement , x Idents(sub_obj)) # 打印当前物种包含的细胞类型方便监控进度 cts - levels(Idents(sub_obj)) cat( 该物种包含, length(cts), 类细胞正在计算...\n) # FindAllMarkers 在 plan(sequential) 下会单核运行 markers - FindAllMarkers( sub_obj, only.pos TRUE, min.pct 0.1, logfc.threshold 0, return.thresh 1 ) if (nrow(markers) 0) { # 每个 Cluster 提取 Top 2000 markers_top2000 - markers %% group_by(cluster) %% slice_max(n 2000, order_by avg_log2FC, with_ties FALSE) %% ungroup() %% mutate(cluster paste0(cluster, _, sp)) all_markers_list[[sp]] - markers_top2000 cat(✅ 物种, sp, 处理完毕已提取 Top 2000。\n) } # ️ 关键手动清理子集内存防止累积崩溃 rm(sub_obj) gc() } # 3. 合并导出 final_deg_df - do.call(rbind, all_markers_list) setwd(/data/work/ldsc_input/a1/) write.csv(final_deg_df, a1_top2000_celltype_markers2human.csv, row.names FALSE) cat(\n [单线程模式] 任务全部完成\n)生成bed文件根据下载的gtf文件library(dplyr) library(rtracklayer) # 路径配置区 setwd(/data/work/ldsc_input/a1/) deg_file - /data/work/ldsc_input/a1/a1_all_celltype_markers2human.csv # 填入你这绝对正确的 v19 版本 GTF 路径 gtf_file - /data/work/xiangmu/cankao/gencode.v19.annotation.gtf # # 1. 读取 DEG 数据并进行严格过滤 (保证基因的特异性) DEG_df - read.csv(deg_file) DEG_df - DEG_df %% filter(p_val_adj 0.05 avg_log2FC 0.5) # 2. 读取并处理 GTF 文件 (v19 文件比较大加载需要一会儿) cat(正在加载 GENCODE v19 GTF 文件...\n) gtf - rtracklayer::import(gtf_file) gtf - gtf[gtf$type gene] gtf_df - as.data.frame(gtf) # 3. 创建输出 BED 文件的文件夹 out_dir - 01.celtype.DEG.star.end dir.create(out_dir, recursive TRUE, showWarnings FALSE) # 4. 循环生成每个细胞类型的 BED 文件 clusters - unique(DEG_df$cluster) cat(\n开始提取 hg19 物理坐标并生成 BED 文件...\n) for (ct in clusters) { # 提取基因 celltype_genes - DEG_df %% filter(cluster ct) %% pull(gene) # 在 v19 GTF 中查找对应的 hg19 物理坐标 nc_gtf - gtf_df %% filter(gene_name %in% celltype_genes) bed_data - nc_gtf[, c(seqnames, start, end)] if(nrow(bed_data) 0) { # 净化文件名 safe_ct - gsub([ /.-], _, ct) safe_ct - gsub(_, _, safe_ct) out_file - file.path(out_dir, paste0(safe_ct, .bed)) write.table(bed_data, out_file, quote FALSE, row.names FALSE, col.names FALSE, sep \t) } else { cat(⚠️ 警告:, ct, 没有在 v19 GTF 中匹配到坐标\n) } } cat(\n 大功告成所有 BED 文件已完美生成\n)library(dplyr) library(rtracklayer) # 路径配置区 setwd(/data/work/ldsc_input/a1/) deg_file - /data/work/ldsc_input/a1/a1_all_celltype_markers2human.csv # GENCODE v19 (hg19) gtf_file - /data/work/xiangmu/cankao/gencode.v19.annotation.gtf # # 1. 读取 DEG 数据 DEG_df - read.csv(deg_file) # 只保留显著 marker DEG_df - DEG_df %% filter(p_val_adj 0.05 avg_log2FC 0.5) # 2. 读取 GTF cat(正在加载 GENCODE v19 GTF 文件...\n) gtf - rtracklayer::import(gtf_file) gtf - gtf[gtf$type gene] gtf_df - as.data.frame(gtf) # 只保留需要的列 gtf_df - gtf_df %% select(seqnames, start, end, gene_name) # 3. 创建输出目录 out_dir - 01.celtype.DEG.star.end dir.create(out_dir, recursive TRUE, showWarnings FALSE) # 4. 获取所有 celltype clusters - unique(DEG_df$cluster) cat(\n开始生成 BED 文件...\n) for (ct in clusters) { cat(处理:, ct, \n) # 提取该细胞类型基因 celltype_genes - DEG_df %% filter(cluster ct) %% pull(gene) # GTF 取交集 nc_gtf - gtf_df %% filter(gene_name %in% celltype_genes) if(nrow(nc_gtf) 0){ cat(⚠️ 警告:, ct, 没有匹配到基因坐标\n) next } # 提取 BED 信息 bed_data - nc_gtf %% select(seqnames, start, end) %% distinct() # 关键修正1chr1 - 1 bed_data$seqnames - gsub(^chr,,bed_data$seqnames) # 关键修正2BED 使用 0-based start bed_data$start - bed_data$start - 1 # 清理文件名 safe_ct - gsub([ /.-], *, ct) safe_ct - gsub(*, _, safe_ct) out_file - file.path(out_dir, paste0(safe_ct, .bed)) write.table( bed_data, out_file, quote FALSE, row.names FALSE, col.names FALSE, sep \t ) } cat(\n 所有 BED 文件生成完成\n)要先给bed文件染色体列加上chr然后再生成annot文件cd /data/work/ldsc_input/a1/01.celtype.DEG.star.end_1500_38_100k/ # 批量给所有 BED 文件第一列加上 chr sed -i s/^/chr/g *.bed # 检查一下确保是 chr1, chr12 这种格式 head -n 5 Amygdala_excitatory_3.bed生成annot文件#!/usr/bin/bash # 路径配置区 (已更新) # 1. 你克隆的 LDSC 目录 LDSC_PATH/data/work/ldsc_input/ldsc/ldsc/ # 2. 你的细胞类型 BED 文件所在目录 BED_DIR/data/work/ldsc_input/a1/01.celtype.DEG.star.end/ # 3. 【关键更新】官方新版参考文件目录 # 注意确保这里指向你解压出的 *.bim 文件所在的文件夹 NEW_REF_DIR/data/work/EUR_REF/1000G_EUR_Phase3_plink # 4. 结果输出目录 (建议新开一个目录防止和旧的混淆) OUT_DIR/data/work/ldsc_input/a1/annot_new_ref mkdir -p ${OUT_DIR} CONDA_BASE$(conda info --base) # source ${CONDA_BASE}/etc/profile.d/conda.sh conda activate ldsc echo 开始使用官方 1000G Phase3 面板生成注释文件... cd ${BED_DIR} for bed in *.bed; do # 提取细胞类型名称 ct_name$(basename ${bed} .bed) echo ------------------------------------------------ echo ⏳ 正在处理: ${ct_name} for j in {1..22}; do # 这里使用了新版 1000G 的命名规则1000G.EUR.QC.${j}.bim python ${LDSC_PATH}/make_annot.py \ --bed-file ${bed} \ --bimfile ${NEW_REF_DIR}/1000G.EUR.QC.${j}.bim \ --annot-file ${OUT_DIR}/${ct_name}.${j}.annot.gz done echo ✅ ${ct_name} 完成 done echo 所有 Annot 文件已生成在: ${OUT_DIR}按物种和细胞类型写ld score的sh脚本这会把每个物种每个细胞类型单独计算#!/usr/bin/bash # 路径配置 BASE_DIR/data/work/ldsc_input LDSC_PY${BASE_DIR}/ldsc/ldsc/ldsc.py ANNOT_DIR${BASE_DIR}/a1/annot_new_ref NEW_BIM_PREFIX/data/work/EUR_REF/1000G_EUR_Phase3_plink/1000G.EUR.QC LDSC_OUT${BASE_DIR}/a1/ldscore_new_ref SCRIPT_OUT${BASE_DIR}/a1/scripts_new_ref mkdir -p ${LDSC_OUT} mkdir -p ${SCRIPT_OUT} CONDA_BASE$(conda info --base) # echo 正在生成【官方面板 Thin Annot 适配版】的子任务脚本... cd ${ANNOT_DIR} for annot_file in *.1.annot.gz; do ct$(basename ${annot_file} .1.annot.gz) job_file${SCRIPT_OUT}/run_ldsc_${ct}.sh cat ${job_file} -EOF #!/usr/bin/bash source ${CONDA_BASE}/etc/profile.d/conda.sh conda activate ldsc echo --- 正在计算 (Thin Annot 模式): ${ct} --- for j in {1..22} do # 关键点因为你的 Annot 是 thin 格式必须保留 --thin-annot python ${LDSC_PY} \\ --l2 \\ --bfile ${NEW_BIM_PREFIX}.\${j} \\ --ld-wind-cm 1 \\ --annot ${ANNOT_DIR}/${ct}.\${j}.annot.gz \\ --thin-annot \\ --out ${LDSC_OUT}/${ct}.\${j} done echo ✅ ${ct} LD Score 计算完成 EOF chmod x ${job_file} done echo 脚本生成完毕已经为您适配了 Thin 格式。离线任务投递ls /data/work/ldsc_input/a1/scripts_new_ref_2000/*.sh | xargs -I {} -P 5 bash {}对gwas文件进行处理先把他转换成带p值的数据python - EOF import pandas as pd import numpy as np import os from scipy.stats import norm import glob in_dir /data/users/duheyang1/duheyang1_68a6ec8cc93b4ae699a7ef3d191e0122/online/GWAS_sort/ out_dir /data/work/ldsc_input/gwas_p/ os.makedirs(out_dir, exist_okTrue) files glob.glob(in_dir *.sumstats.gz) print(f发现 {len(files)} 个GWAS文件) for f in files: name os.path.basename(f) out os.path.join(out_dir, name) print(处理:, name) df pd.read_csv(f, sep\t) if Z not in df.columns: print(跳过 (没有Z列):, name) continue # Z - P df[P] 2 * norm.sf(np.abs(df[Z])) df.to_csv(out, sep\t, indexFalse, compressiongzip) print(全部完成) EOF然后整理成ldsc输入数据格式#!/usr/bin/bash echo 转换 [P Z 校验模式] source $(conda info --base)/etc/profile.d/conda.sh conda activate ldsc MUNGE/data/work/ldsc_input/ldsc/ldsc/munge_sumstats.py GWAS_DIR/data/work/ldsc_input/gwas_p OUT_DIR/data/work/ldsc_input/munged_sumstats HM3_SNPLIST/data/work/EUR_REF/w_hm3.snplist mkdir -p ${OUT_DIR} for gwas in ${GWAS_DIR}/*.gz do base$(basename ${gwas}) # 适配不同的后缀名提取 name${base%.sumstats.gz} name${name%.gz} echo -------------------------------------------- echo 正在处理: ${name} python ${MUNGE} \ --sumstats ${gwas} \ --merge-alleles ${HM3_SNPLIST} \ --snp SNP \ --a1 A1 \ --a2 A2 \ --p P \ --N-col N \ --signed-sumstats Z,0 \ --chunksize 500000 \ --out ${OUT_DIR}/${name} echo ✅ ${name} 转换完成 done echo 所有 GWAS 处理成功生成按细胞类型的脚本文件#!/usr/bin/env bash # 定义新的路径 BASE_DIR/data/work/ldsc_input/a1 NEW_LDSCORE_DIR${BASE_DIR}/ldscore_new_ref NEW_CTS_CONFIG_DIR${BASE_DIR}/cts_config_new_ref # 创建输出目录 mkdir -p ${NEW_CTS_CONFIG_DIR} echo 开始生成新的细胞类型映射文件 (.ldsc)... # 进入新的 ldscore 目录 cd ${NEW_LDSCORE_DIR} || { echo ❌ 无法进入目录: ${NEW_LDSCORE_DIR}; exit 1; } # 遍历所有符合 LDSC 命名规范的文件 # 注意这里假设你的文件名后缀依然是 .1.l2.ldscore.gz for file in *.1.l2.ldscore.gz do # 提取细胞类型名称 (去掉后缀) ct$(basename ${file} .1.l2.ldscore.gz) # 生成映射内容细胞类型名称 路径前缀带点号 # 结果保存到新的 cts_config 目录下 echo ${ct} ${NEW_LDSCORE_DIR}/${ct}. ${NEW_CTS_CONFIG_DIR}/${ct}.ldsc echo ✅ 生成成功: ${ct}.ldsc done echo 恭喜所有映射文件已保存在: ${NEW_CTS_CONFIG_DIR}回归分析#!/usr/bin/env bash # 0. 激活 Conda 环境 echo 正在激活 ldsc 环境... CONDA_BASE$(conda info --base) source ${CONDA_BASE}/etc/profile.d/conda.sh conda activate ldsc # 1. 基础工具与参考面板路径 LDSC_PY/data/work/ldsc_input/ldsc/ldsc/ldsc.py # 更新为 baseline_v1.2 路径 BASELINE_DIR/data/work/EUR_REF/baseline_v1.2 WEIGHT_DIR/data/work/EUR_REF/1000G_Phase3_weights_hm3_no_MHC # 2. 你的新数据路径 # 更新 GWAS 文件路径 SUMSTATS_DIR/data/work/ldsc_input/munged_sumstats # 更新新生成的映射文件路径 CTS_CONFIG_DIR/data/work/ldsc_input/a1/cts_config_new_ref # 更新结果输出目录 RESULT_DIR/data/work/ldsc_input/a1/regression_results_new_ref mkdir -p ${RESULT_DIR} # 3. 并发控制 MAX_THREADS8 echo 开始执行新参考面板下的 LDSC 细胞特异性分析 (线程限制: ${MAX_THREADS})... # 遍历 GWAS 文件 for sumstat in ${SUMSTATS_DIR}/*.sumstats.gz; do disease$(basename ${sumstat} .sumstats.gz) echo echo 处理性状: ${disease} # 遍历细胞类型配置文件 for cts_file in ${CTS_CONFIG_DIR}/*.ldsc; do ct_name$(basename ${cts_file} .ldsc) # 运行 LDSC 回归 # 注意--ref-ld-chr 使用了 v1.2 的前缀 baseline. python ${LDSC_PY} \ --h2-cts ${sumstat} \ --ref-ld-chr ${BASELINE_DIR}/baseline. \ --out ${RESULT_DIR}/${disease}_${ct_name} \ --ref-ld-chr-cts ${cts_file} \ --w-ld-chr ${WEIGHT_DIR}/weights.hm3_noMHC. /dev/null 21 # 线程并发控制逻辑 while [ $(jobs -p | wc -l) -ge ${MAX_THREADS} ]; do sleep 1 done done done # 等待收尾 echo ⏳ 任务投递完成等待后台进程结束... wait echo 全部分析任务已完成结果请查看: ${RESULT_DIR}
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.coloradmin.cn/o/2566630.html
如若内容造成侵权/违法违规/事实不符,请联系多彩编程网进行投诉反馈,一经查实,立即删除!相关文章
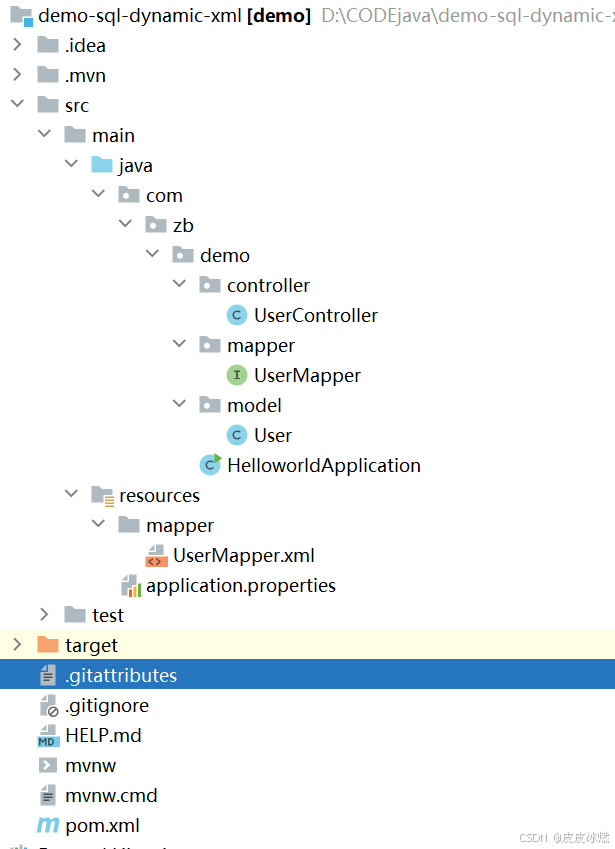
SpringBoot-17-MyBatis动态SQL标签之常用标签
文章目录 1 代码1.1 实体User.java1.2 接口UserMapper.java1.3 映射UserMapper.xml1.3.1 标签if1.3.2 标签if和where1.3.3 标签choose和when和otherwise1.4 UserController.java2 常用动态SQL标签2.1 标签set2.1.1 UserMapper.java2.1.2 UserMapper.xml2.1.3 UserController.ja…
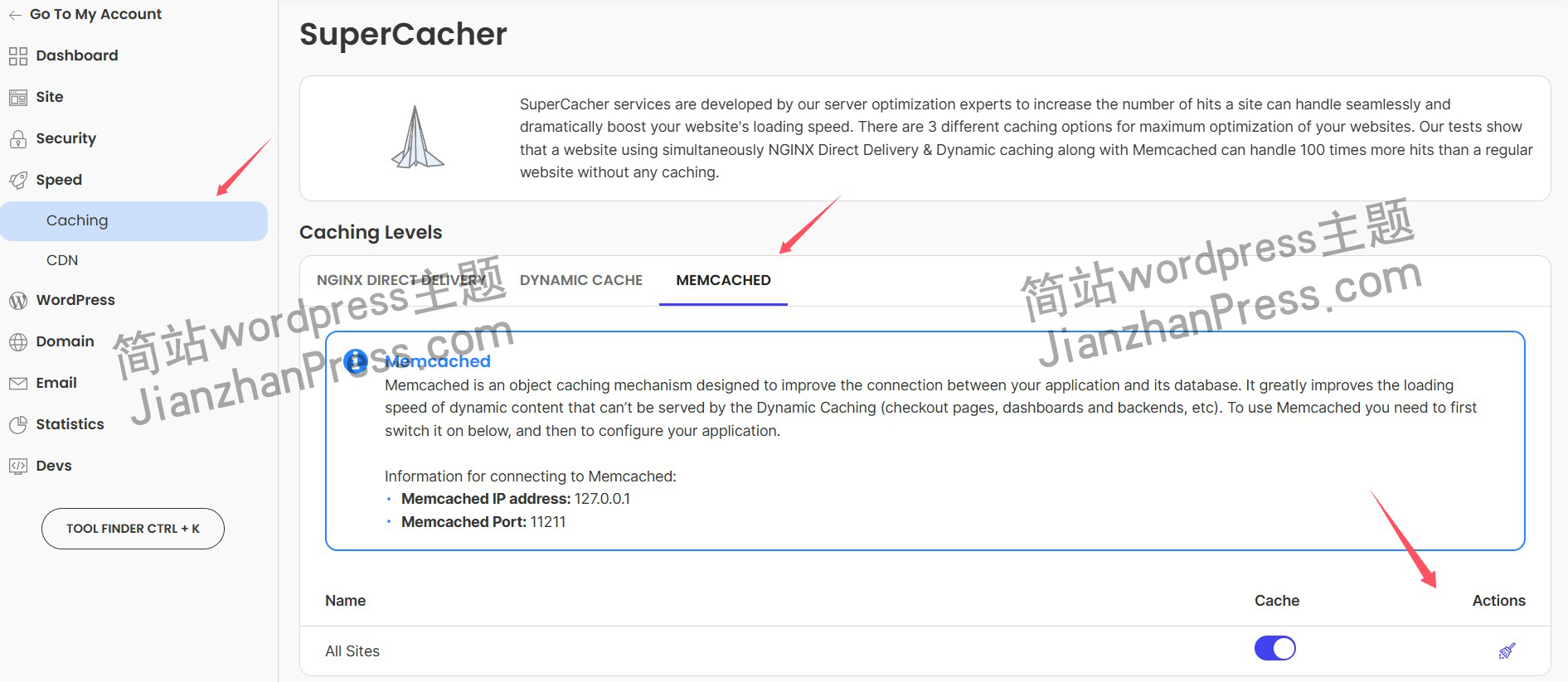
wordpress后台更新后 前端没变化的解决方法
使用siteground主机的wordpress网站,会出现更新了网站内容和修改了php模板文件、js文件、css文件、图片文件后,网站没有变化的情况。
不熟悉siteground主机的新手,遇到这个问题,就很抓狂,明明是哪都没操作错误&#x…
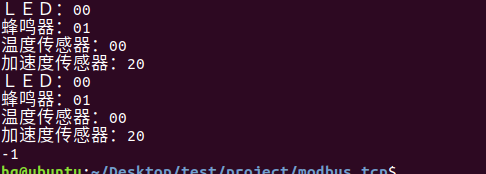
网络编程(Modbus进阶)
思维导图 Modbus RTU(先学一点理论)
概念 Modbus RTU 是工业自动化领域 最广泛应用的串行通信协议,由 Modicon 公司(现施耐德电气)于 1979 年推出。它以 高效率、强健性、易实现的特点成为工业控制系统的通信标准。 包…
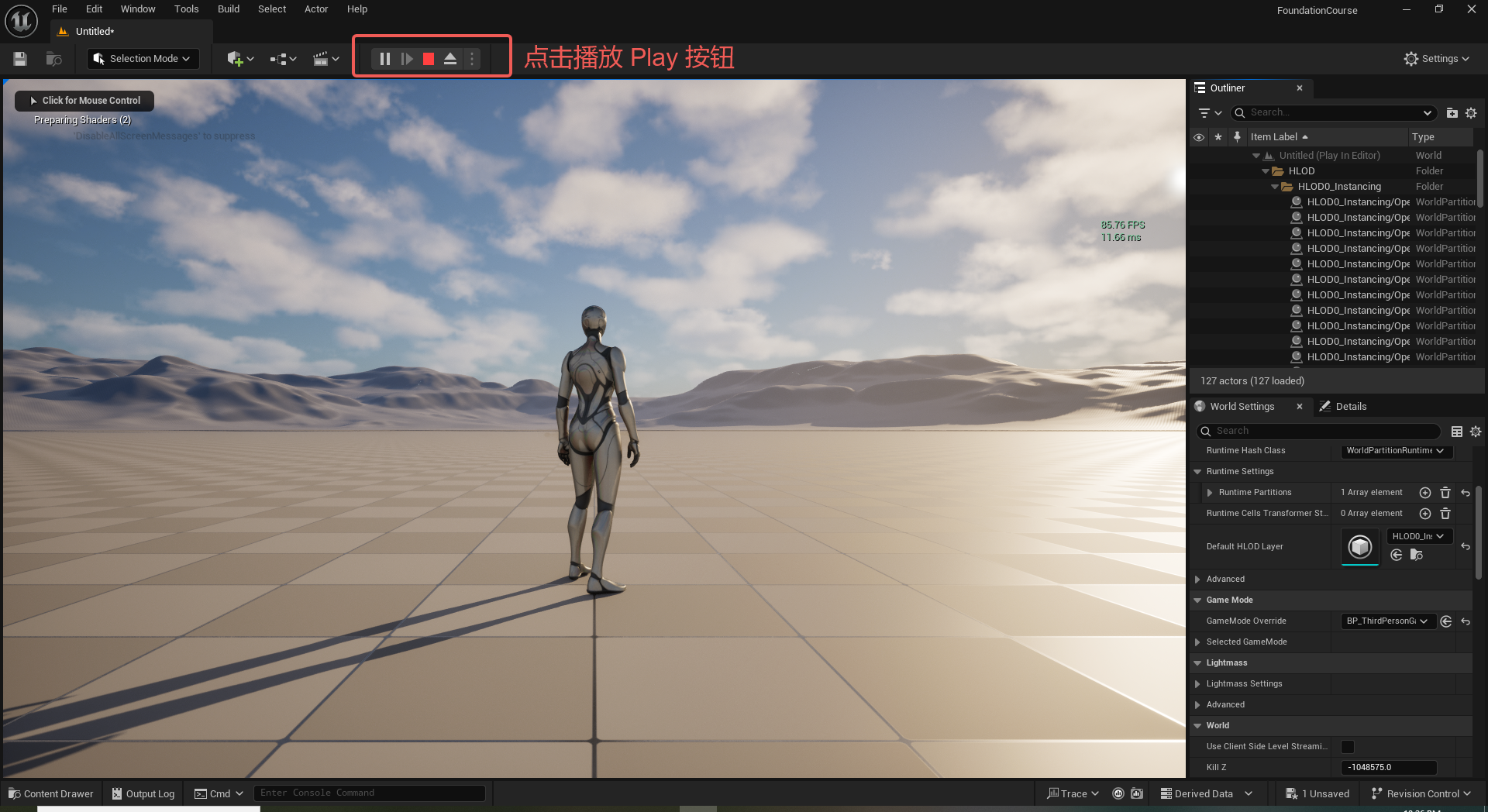
UE5 学习系列(二)用户操作界面及介绍
这篇博客是 UE5 学习系列博客的第二篇,在第一篇的基础上展开这篇内容。博客参考的 B 站视频资料和第一篇的链接如下:
【Note】:如果你已经完成安装等操作,可以只执行第一篇博客中 2. 新建一个空白游戏项目 章节操作,重…
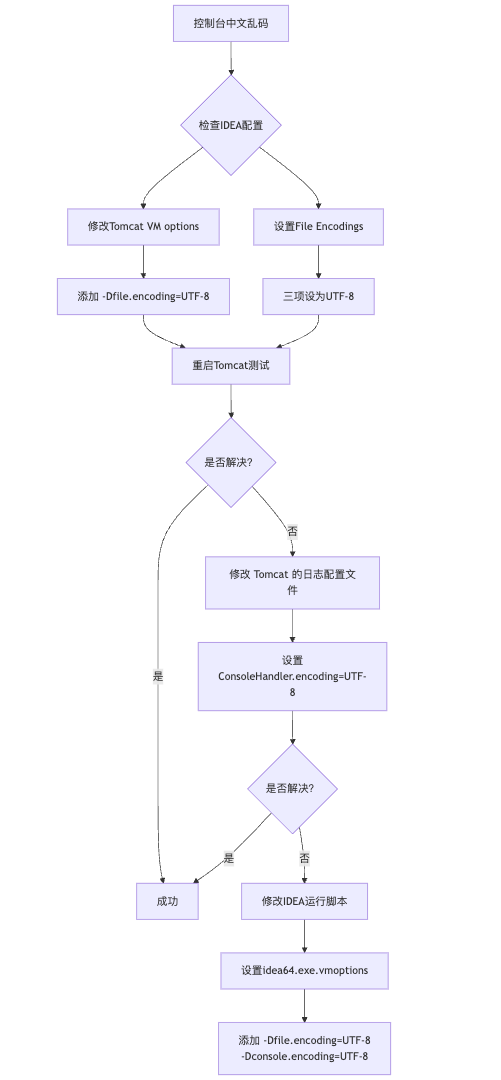
IDEA运行Tomcat出现乱码问题解决汇总
最近正值期末周,有很多同学在写期末Java web作业时,运行tomcat出现乱码问题,经过多次解决与研究,我做了如下整理:
原因:
IDEA本身编码与tomcat的编码与Windows编码不同导致,Windows 系统控制台…

利用最小二乘法找圆心和半径
#include <iostream>
#include <vector>
#include <cmath>
#include <Eigen/Dense> // 需安装Eigen库用于矩阵运算 // 定义点结构
struct Point { double x, y; Point(double x_, double y_) : x(x_), y(y_) {}
}; // 最小二乘法求圆心和半径 …
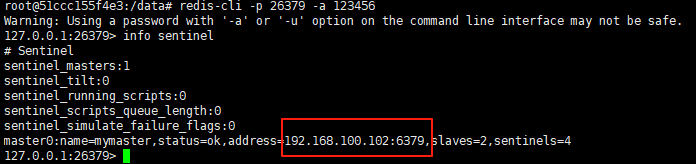
使用docker在3台服务器上搭建基于redis 6.x的一主两从三台均是哨兵模式
一、环境及版本说明
如果服务器已经安装了docker,则忽略此步骤,如果没有安装,则可以按照一下方式安装: 1. 在线安装(有互联网环境): 请看我这篇文章 传送阵>> 点我查看 2. 离线安装(内网环境):请看我这篇文章 传送阵>> 点我查看
说明:假设每台服务器已…
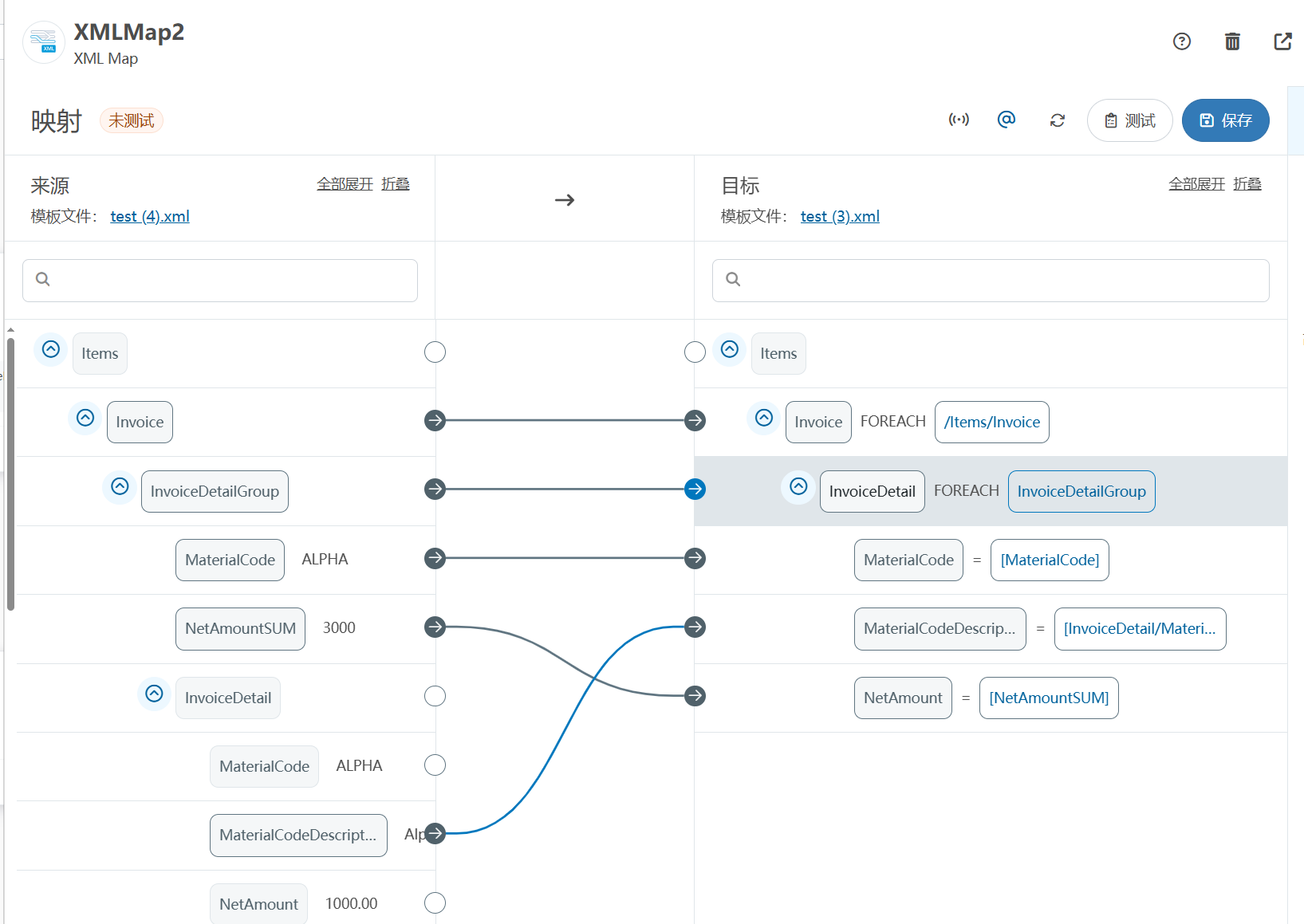
XML Group端口详解
在XML数据映射过程中,经常需要对数据进行分组聚合操作。例如,当处理包含多个物料明细的XML文件时,可能需要将相同物料号的明细归为一组,或对相同物料号的数量进行求和计算。传统实现方式通常需要编写脚本代码,增加了开…
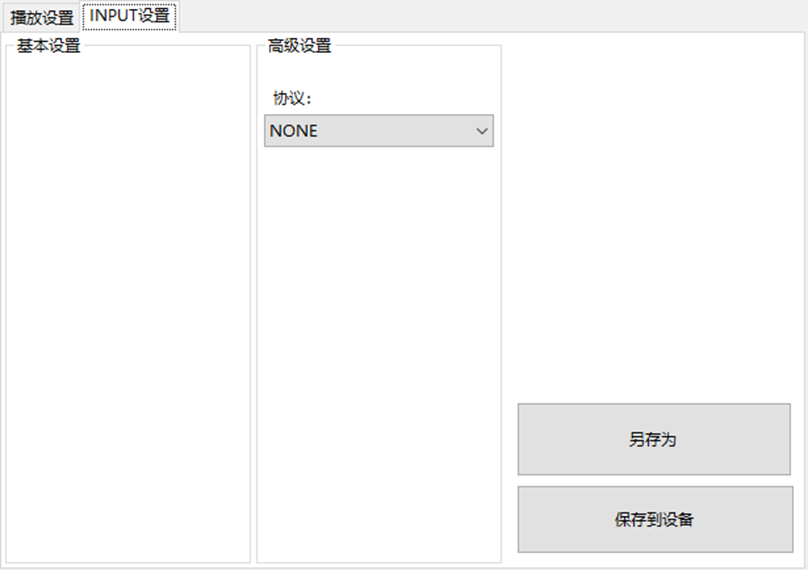
LBE-LEX系列工业语音播放器|预警播报器|喇叭蜂鸣器的上位机配置操作说明
LBE-LEX系列工业语音播放器|预警播报器|喇叭蜂鸣器专为工业环境精心打造,完美适配AGV和无人叉车。同时,集成以太网与语音合成技术,为各类高级系统(如MES、调度系统、库位管理、立库等)提供高效便捷的语音交互体验。
L…

(LeetCode 每日一题) 3442. 奇偶频次间的最大差值 I (哈希、字符串)
题目:3442. 奇偶频次间的最大差值 I 思路 :哈希,时间复杂度0(n)。 用哈希表来记录每个字符串中字符的分布情况,哈希表这里用数组即可实现。
C版本:
class Solution {
public:int maxDifference(string s) {int a[26]…

【大模型RAG】拍照搜题技术架构速览:三层管道、两级检索、兜底大模型
摘要
拍照搜题系统采用“三层管道(多模态 OCR → 语义检索 → 答案渲染)、两级检索(倒排 BM25 向量 HNSW)并以大语言模型兜底”的整体框架: 多模态 OCR 层 将题目图片经过超分、去噪、倾斜校正后,分别用…
【Axure高保真原型】引导弹窗
今天和大家中分享引导弹窗的原型模板,载入页面后,会显示引导弹窗,适用于引导用户使用页面,点击完成后,会显示下一个引导弹窗,直至最后一个引导弹窗完成后进入首页。具体效果可以点击下方视频观看或打开下方…
接口测试中缓存处理策略
在接口测试中,缓存处理策略是一个关键环节,直接影响测试结果的准确性和可靠性。合理的缓存处理策略能够确保测试环境的一致性,避免因缓存数据导致的测试偏差。以下是接口测试中常见的缓存处理策略及其详细说明:
一、缓存处理的核…
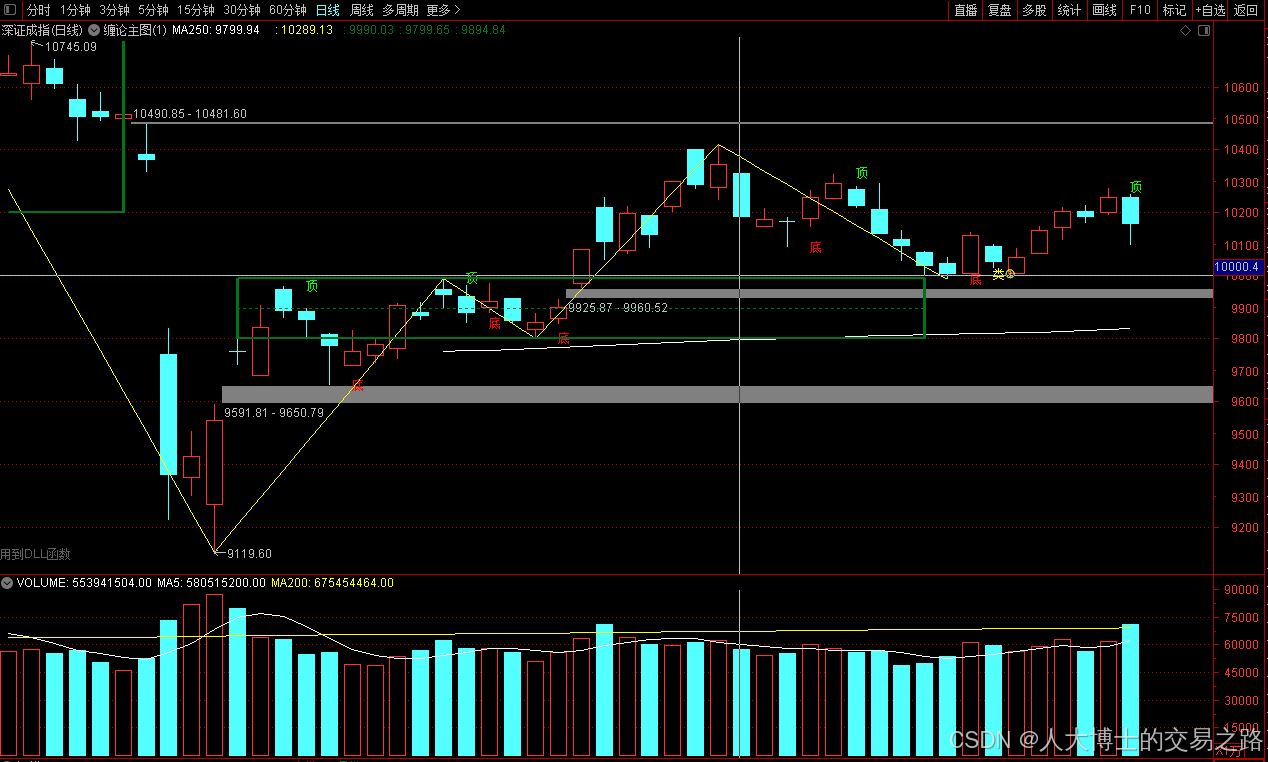
龙虎榜——20250610
上证指数放量收阴线,个股多数下跌,盘中受消息影响大幅波动。 深证指数放量收阴线形成顶分型,指数短线有调整的需求,大概需要一两天。 2025年6月10日龙虎榜行业方向分析 1. 金融科技
代表标的:御银股份、雄帝科技
驱动…
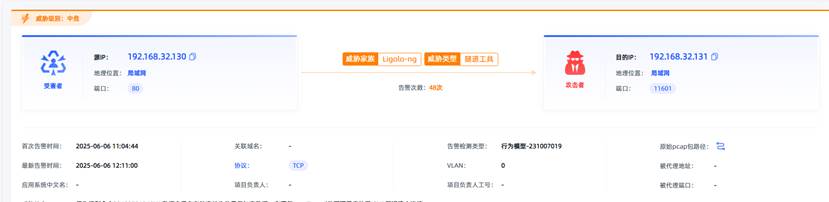
观成科技:隐蔽隧道工具Ligolo-ng加密流量分析
1.工具介绍
Ligolo-ng是一款由go编写的高效隧道工具,该工具基于TUN接口实现其功能,利用反向TCP/TLS连接建立一条隐蔽的通信信道,支持使用Let’s Encrypt自动生成证书。Ligolo-ng的通信隐蔽性体现在其支持多种连接方式,适应复杂网…

铭豹扩展坞 USB转网口 突然无法识别解决方法
当 USB 转网口扩展坞在一台笔记本上无法识别,但在其他电脑上正常工作时,问题通常出在笔记本自身或其与扩展坞的兼容性上。以下是系统化的定位思路和排查步骤,帮助你快速找到故障原因:
背景:
一个M-pard(铭豹)扩展坞的网卡突然无法识别了,扩展出来的三个USB接口正常。…

未来机器人的大脑:如何用神经网络模拟器实现更智能的决策?
编辑:陈萍萍的公主一点人工一点智能 未来机器人的大脑:如何用神经网络模拟器实现更智能的决策?RWM通过双自回归机制有效解决了复合误差、部分可观测性和随机动力学等关键挑战,在不依赖领域特定归纳偏见的条件下实现了卓越的预测准…

Linux应用开发之网络套接字编程(实例篇)
服务端与客户端单连接
服务端代码
#include <sys/socket.h>
#include <sys/types.h>
#include <netinet/in.h>
#include <stdio.h>
#include <stdlib.h>
#include <string.h>
#include <arpa/inet.h>
#include <pthread.h>
…

华为云AI开发平台ModelArts
华为云ModelArts:重塑AI开发流程的“智能引擎”与“创新加速器”!
在人工智能浪潮席卷全球的2025年,企业拥抱AI的意愿空前高涨,但技术门槛高、流程复杂、资源投入巨大的现实,却让许多创新构想止步于实验室。数据科学家…

深度学习在微纳光子学中的应用
深度学习在微纳光子学中的主要应用方向
深度学习与微纳光子学的结合主要集中在以下几个方向:
逆向设计 通过神经网络快速预测微纳结构的光学响应,替代传统耗时的数值模拟方法。例如设计超表面、光子晶体等结构。
特征提取与优化 从复杂的光学数据中自…