Visium HD空转实战:Space Ranger v4.0.1从安装到结果解读全流程
news2026/4/2 4:05:07
1. Visium HD与Space Ranger初探第一次接触Visium HD技术时我被它强大的空间转录组分析能力震撼到了。简单来说这项技术能让我们在组织切片上精确到单个细胞的位置同时获取它们的基因表达数据。想象一下这就像给组织样本拍了一张高清照片同时还能知道每个像素点里发生了什么分子事件。Space Ranger是10x Genomics官方提供的分析软件最新版本v4.0.1针对Visium HD做了专门优化。我实测下来发现相比旧版本它在处理高清数据时速度提升了约30%内存占用也更为合理。对于刚接触这个领域的研究人员掌握Space Ranger的使用是开展空间转录组研究的必备技能。这个实战指南会带你走完从软件安装到结果解读的全过程。即使你没有任何生物信息学背景跟着我的步骤操作也能在一天内完成整个分析流程。我特别整理了新手最容易出错的几个环节比如环境变量设置和参数选择帮你避开那些我当年踩过的坑。2. 环境准备与软件安装2.1 系统要求检查在开始安装前务必确认你的服务器或工作站满足最低配置要求。根据我的经验处理Visium HD数据至少需要64位Linux系统CentOS 7或Ubuntu 18.0464GB内存推荐128GB以上500GB可用存储空间16核CPU可以用这些命令快速检查你的系统配置# 查看内存 free -h # 查看CPU核心数 nproc # 查看磁盘空间 df -h2.2 下载与安装Space Ranger10x Genomics官方提供了压缩包形式的软件包。我建议直接使用curl下载避免浏览器下载可能出现的网络中断问题curl -o spaceranger-4.0.1.tar.gz 下载链接 tar -xzvf spaceranger-4.0.1.tar.gz解压后会得到一个名为spaceranger-4.0.1的目录。这里有个小技巧我习惯把软件安装在/project/Software/目录下方便团队其他成员使用。2.3 环境变量配置为了让系统能识别spaceranger命令需要将其添加到PATH环境变量中echo export PATH$PATH:/project/Software/spaceranger-4.0.1 ~/.bashrc source ~/.bashrc验证安装是否成功spaceranger --version如果看到spaceranger 4.0.1的输出说明安装正确。我在第一次安装时忘了source命令结果怎么都找不到spaceranger折腾了半天才发现问题。3. 参考基因组与探针集准备3.1 下载参考基因组Space Ranger分析需要物种特异的参考基因组。以人类GRCh38为例curl -O https://cf.10xgenomics.com/supp/spatial-exp/refdata-gex-GRCh38-2020-A.tar.gz tar -xzvf refdata-gex-GRCh38-2020-A.tar.gz这个参考基因组大约有30GB下载时间取决于你的网络速度。我建议在服务器上用screen或tmux运行避免SSH断开导致下载中断。3.2 获取探针集文件Visium HD使用特制的探针集需要从10x Genomics官网下载。选择与你参考基因组匹配的版本很重要。比如GRCh38参考基因组就要配GRCh38探针集。下载后检查文件完整性md5sum Visium_Human_Transcriptome_Probe_Set_v2.0_GRCh38-2020-A.csv应该与官网提供的MD5值一致。我曾经遇到过文件损坏导致分析失败的情况现在每次都习惯性做这个检查。4. 数据准备与质量检查4.1 FASTQ文件组织测序公司通常会提供压缩的FASTQ文件。解压后目录结构应该是这样的fastq_dir/ ├── sample_S1_L001_R1_001.fastq.gz ├── sample_S1_L001_R2_001.fastq.gz ├── sample_S1_L002_R1_001.fastq.gz └── sample_S1_L002_R2_001.fastq.gz建议使用tree命令快速查看目录结构tree -L 2 fastq_dir4.2 图像文件准备Visium HD分析需要组织切片的图像文件。常见格式有.tif用于--cytaimage参数.btf用于--image参数我建议同时准备两种格式因为某些下游分析工具可能有特定要求。检查图像文件是否完整file CAVG10539_2023-11-16_14-56-24_APPS115_H1-YD7CDZK_A1_S11088.tif5. 运行Space Ranger count5.1 参数详解spaceranger count是核心分析命令关键参数包括--id输出目录名称--transcriptome参考基因组路径--fastqsFASTQ文件目录--probe-set探针集文件路径--slide玻片编号--area捕获区域编号--cytaimage组织图像文件--create-bam是否输出BAM文件5.2 实际运行示例这是一个完整的运行命令spaceranger count --idhd_count \ --transcriptome/path/to/refdata-gex-GRCh38-2020-A \ --fastqs/path/to/fastq \ --probe-set/path/to/Visium_Human_Transcriptome_Probe_Set_v2.0_GRCh38-2020-A.csv \ --slideH1-YD7CDZK \ --areaA1 \ --cytaimage/path/to/CAVG10539_2023-11-16_14-56-24_APPS115_H1-YD7CDZK_A1_S11088.tif \ --image/path/to/APPS115_11088_rescan_01.btf \ --create-bamtrue运行时间取决于数据量通常需要6-12小时。建议使用nohup或screen保持会话nohup spaceranger count ... count.log 21 5.3 监控运行状态可以通过以下方式监控进度tail -f hd_count/_log或者查看资源使用情况top -u your_username6. 结果解读与质量控制6.1 输出文件结构成功运行后会生成以下重要文件web_summary.html质量报告spatial/空间数据cloupe.cloupeLoupe Browser可视化文件barcodes.tsv.gz细胞条形码6.2 解读web_summary.html这是最重要的质量报告重点关注测序指标总reads数有效reads比例Q30分值空间捕获指标组织覆盖率中位基因数/点中位UMI数/点探针结合效率探针结合reads比例目标区域覆盖度我通常会把这些指标整理成表格方便多个样本间比较。如果发现有效reads比例低于60%可能需要重新检查实验步骤。6.3 常见问题排查低映射率检查参考基因组是否匹配高重复率可能是PCR扩增过度低探针效率检查探针集版本组织覆盖不均可能是样本处理问题7. 下游分析准备7.1 数据导出Space Ranger的输出可以直接用于多种下游分析工具Seurat单细胞分析ScanpyPython分析流程Giotto空间分析专用工具我通常先用Loupe Browser快速查看空间分布再用Seurat进行深入分析。7.2 自定义bin大小Visium HD支持自定义空间bin大小--custom-bin-size 5这个功能在研究特定组织结构时特别有用比如肿瘤微环境中不同区域的基因表达差异。7.3 多样本整合如果有多个Visium HD样本可以使用spaceranger aggr命令合并分析。记得提前准备好aggregation.csv文件指定各样本路径和比例。在实际项目中我发现先单独分析每个样本再用Seurat的整合方法效果更好。这能保留更多样本特异性信息特别是在处理异质性较强的组织时。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.coloradmin.cn/o/2474148.html
如若内容造成侵权/违法违规/事实不符,请联系多彩编程网进行投诉反馈,一经查实,立即删除!相关文章
SpringBoot-17-MyBatis动态SQL标签之常用标签
文章目录 1 代码1.1 实体User.java1.2 接口UserMapper.java1.3 映射UserMapper.xml1.3.1 标签if1.3.2 标签if和where1.3.3 标签choose和when和otherwise1.4 UserController.java2 常用动态SQL标签2.1 标签set2.1.1 UserMapper.java2.1.2 UserMapper.xml2.1.3 UserController.ja…
wordpress后台更新后 前端没变化的解决方法
使用siteground主机的wordpress网站,会出现更新了网站内容和修改了php模板文件、js文件、css文件、图片文件后,网站没有变化的情况。
不熟悉siteground主机的新手,遇到这个问题,就很抓狂,明明是哪都没操作错误&#x…
网络编程(Modbus进阶)
思维导图 Modbus RTU(先学一点理论)
概念 Modbus RTU 是工业自动化领域 最广泛应用的串行通信协议,由 Modicon 公司(现施耐德电气)于 1979 年推出。它以 高效率、强健性、易实现的特点成为工业控制系统的通信标准。 包…
UE5 学习系列(二)用户操作界面及介绍
这篇博客是 UE5 学习系列博客的第二篇,在第一篇的基础上展开这篇内容。博客参考的 B 站视频资料和第一篇的链接如下:
【Note】:如果你已经完成安装等操作,可以只执行第一篇博客中 2. 新建一个空白游戏项目 章节操作,重…
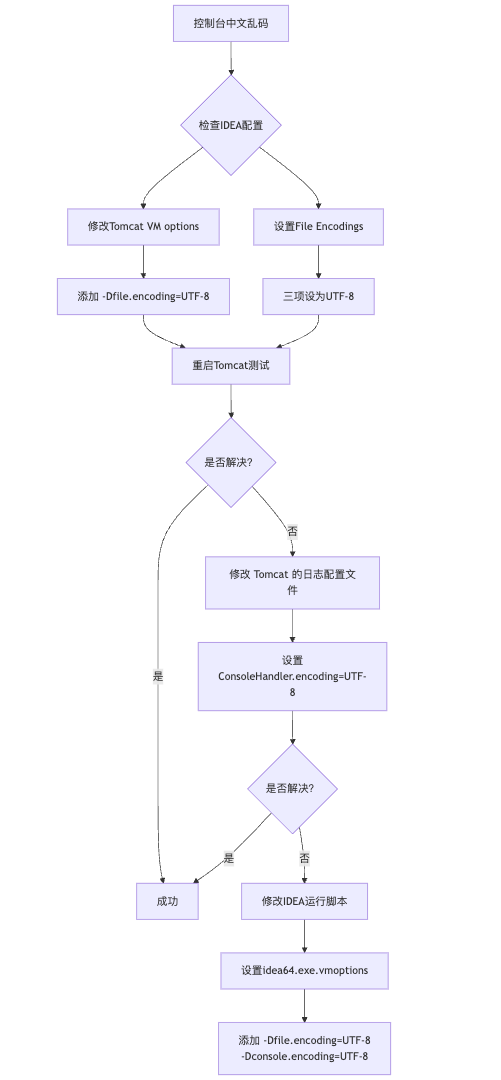
IDEA运行Tomcat出现乱码问题解决汇总
最近正值期末周,有很多同学在写期末Java web作业时,运行tomcat出现乱码问题,经过多次解决与研究,我做了如下整理:
原因:
IDEA本身编码与tomcat的编码与Windows编码不同导致,Windows 系统控制台…

利用最小二乘法找圆心和半径
#include <iostream>
#include <vector>
#include <cmath>
#include <Eigen/Dense> // 需安装Eigen库用于矩阵运算 // 定义点结构
struct Point { double x, y; Point(double x_, double y_) : x(x_), y(y_) {}
}; // 最小二乘法求圆心和半径 …
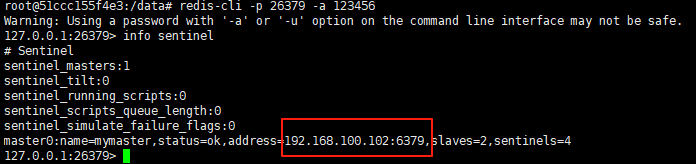
使用docker在3台服务器上搭建基于redis 6.x的一主两从三台均是哨兵模式
一、环境及版本说明
如果服务器已经安装了docker,则忽略此步骤,如果没有安装,则可以按照一下方式安装: 1. 在线安装(有互联网环境): 请看我这篇文章 传送阵>> 点我查看 2. 离线安装(内网环境):请看我这篇文章 传送阵>> 点我查看
说明:假设每台服务器已…
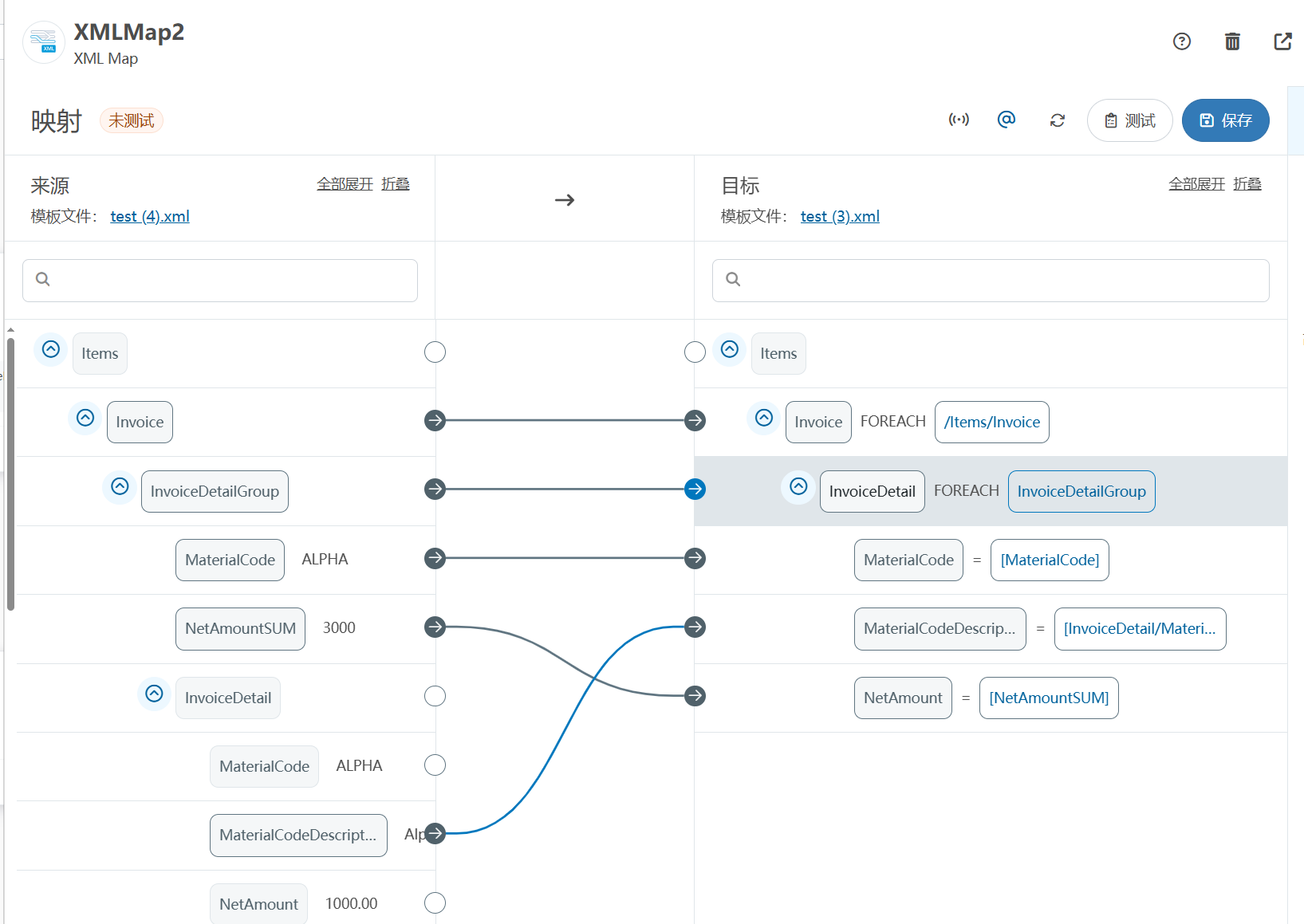
XML Group端口详解
在XML数据映射过程中,经常需要对数据进行分组聚合操作。例如,当处理包含多个物料明细的XML文件时,可能需要将相同物料号的明细归为一组,或对相同物料号的数量进行求和计算。传统实现方式通常需要编写脚本代码,增加了开…
LBE-LEX系列工业语音播放器|预警播报器|喇叭蜂鸣器的上位机配置操作说明
LBE-LEX系列工业语音播放器|预警播报器|喇叭蜂鸣器专为工业环境精心打造,完美适配AGV和无人叉车。同时,集成以太网与语音合成技术,为各类高级系统(如MES、调度系统、库位管理、立库等)提供高效便捷的语音交互体验。
L…

(LeetCode 每日一题) 3442. 奇偶频次间的最大差值 I (哈希、字符串)
题目:3442. 奇偶频次间的最大差值 I 思路 :哈希,时间复杂度0(n)。 用哈希表来记录每个字符串中字符的分布情况,哈希表这里用数组即可实现。
C版本:
class Solution {
public:int maxDifference(string s) {int a[26]…

【大模型RAG】拍照搜题技术架构速览:三层管道、两级检索、兜底大模型
摘要
拍照搜题系统采用“三层管道(多模态 OCR → 语义检索 → 答案渲染)、两级检索(倒排 BM25 向量 HNSW)并以大语言模型兜底”的整体框架: 多模态 OCR 层 将题目图片经过超分、去噪、倾斜校正后,分别用…
【Axure高保真原型】引导弹窗
今天和大家中分享引导弹窗的原型模板,载入页面后,会显示引导弹窗,适用于引导用户使用页面,点击完成后,会显示下一个引导弹窗,直至最后一个引导弹窗完成后进入首页。具体效果可以点击下方视频观看或打开下方…
接口测试中缓存处理策略
在接口测试中,缓存处理策略是一个关键环节,直接影响测试结果的准确性和可靠性。合理的缓存处理策略能够确保测试环境的一致性,避免因缓存数据导致的测试偏差。以下是接口测试中常见的缓存处理策略及其详细说明:
一、缓存处理的核…
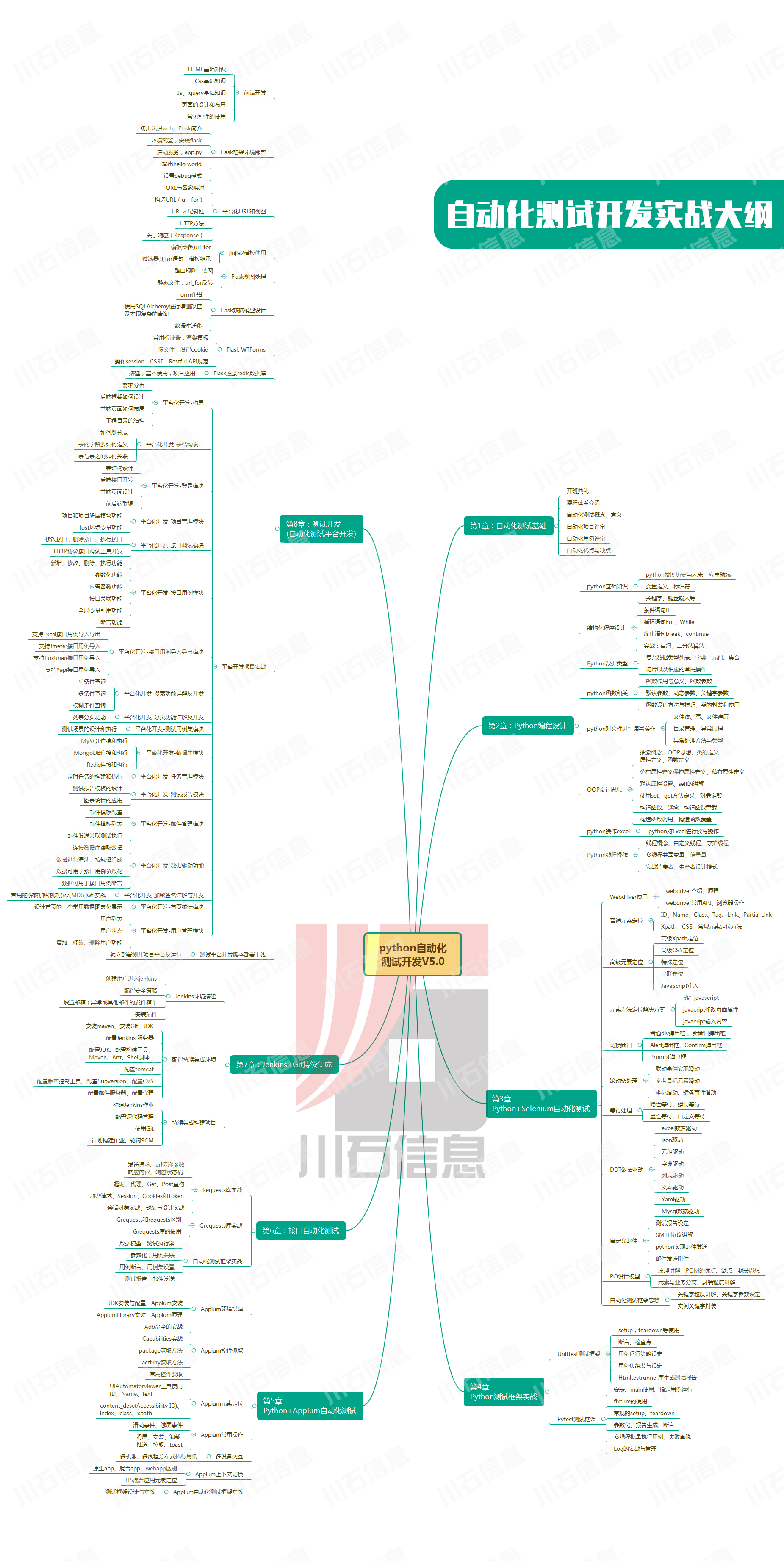
龙虎榜——20250610
上证指数放量收阴线,个股多数下跌,盘中受消息影响大幅波动。 深证指数放量收阴线形成顶分型,指数短线有调整的需求,大概需要一两天。 2025年6月10日龙虎榜行业方向分析 1. 金融科技
代表标的:御银股份、雄帝科技
驱动…
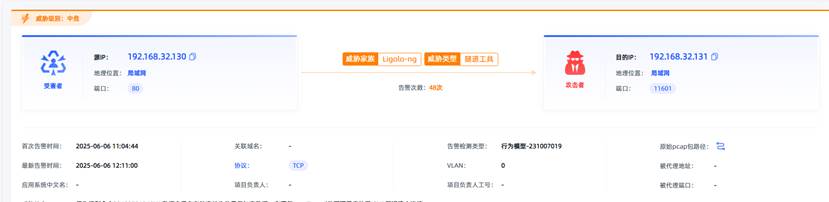
观成科技:隐蔽隧道工具Ligolo-ng加密流量分析
1.工具介绍
Ligolo-ng是一款由go编写的高效隧道工具,该工具基于TUN接口实现其功能,利用反向TCP/TLS连接建立一条隐蔽的通信信道,支持使用Let’s Encrypt自动生成证书。Ligolo-ng的通信隐蔽性体现在其支持多种连接方式,适应复杂网…

铭豹扩展坞 USB转网口 突然无法识别解决方法
当 USB 转网口扩展坞在一台笔记本上无法识别,但在其他电脑上正常工作时,问题通常出在笔记本自身或其与扩展坞的兼容性上。以下是系统化的定位思路和排查步骤,帮助你快速找到故障原因:
背景:
一个M-pard(铭豹)扩展坞的网卡突然无法识别了,扩展出来的三个USB接口正常。…

未来机器人的大脑:如何用神经网络模拟器实现更智能的决策?
编辑:陈萍萍的公主一点人工一点智能 未来机器人的大脑:如何用神经网络模拟器实现更智能的决策?RWM通过双自回归机制有效解决了复合误差、部分可观测性和随机动力学等关键挑战,在不依赖领域特定归纳偏见的条件下实现了卓越的预测准…

Linux应用开发之网络套接字编程(实例篇)
服务端与客户端单连接
服务端代码
#include <sys/socket.h>
#include <sys/types.h>
#include <netinet/in.h>
#include <stdio.h>
#include <stdlib.h>
#include <string.h>
#include <arpa/inet.h>
#include <pthread.h>
…

华为云AI开发平台ModelArts
华为云ModelArts:重塑AI开发流程的“智能引擎”与“创新加速器”!
在人工智能浪潮席卷全球的2025年,企业拥抱AI的意愿空前高涨,但技术门槛高、流程复杂、资源投入巨大的现实,却让许多创新构想止步于实验室。数据科学家…

深度学习在微纳光子学中的应用
深度学习在微纳光子学中的主要应用方向
深度学习与微纳光子学的结合主要集中在以下几个方向:
逆向设计 通过神经网络快速预测微纳结构的光学响应,替代传统耗时的数值模拟方法。例如设计超表面、光子晶体等结构。
特征提取与优化 从复杂的光学数据中自…