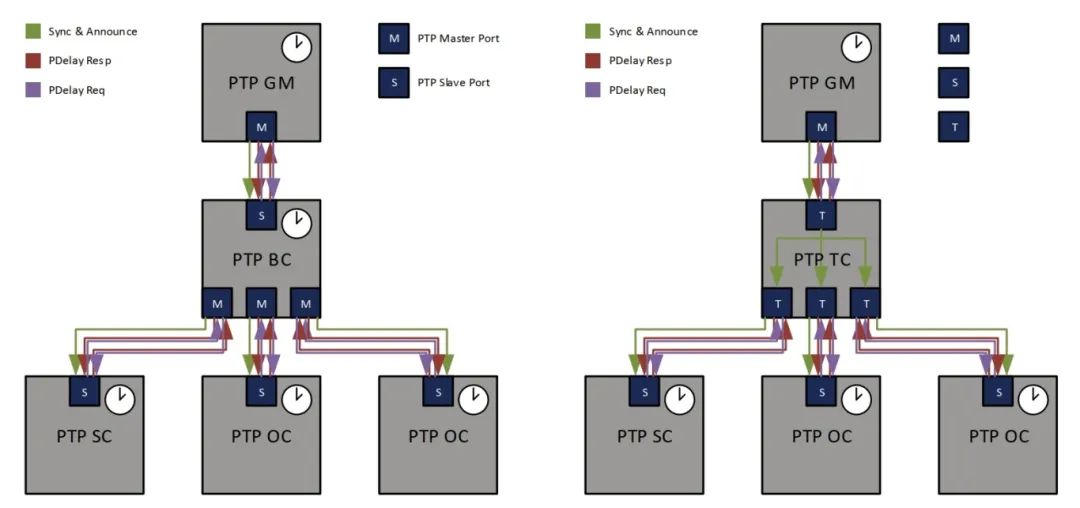
目录
- 方法 1:使用 Ensembl REST API(推荐,适用于少量位点查询)
- 方法 2:使用 UCSC API
- 方法 3:使用 NCBI API 并转换坐标(需要额外步骤)
- 方法 4:使用本地数据库(最可靠,适合批量查询)
- 1、下载 GRCh37 基因注释文件:
- 2、创建 Python 查询脚本:
- 其他
- 注意事项
- Ensembl API 限制:
- 基因组版本差异:
- 基因位置表示:
数据准备:
要获取 GRCh37(hg19)版本的染色体位置,我们需要使用专门针对该基因组版本的数据源。
方法 1:使用 Ensembl REST API(推荐,适用于少量位点查询)
import requests
import time
def get_gene_location_hg19(gene_symbol, max_retries=3):
"""
获取基因在 GRCh37/hg19 版本的位置
"""
base_url = "http://grch37.rest.ensembl.org"
endpoint = f"/lookup/symbol/homo_sapiens/{gene_symbol}"
url = base_url + endpoint
params = {
"expand": 0,
"utr": 0,
"content-type": "application/json"
}
for attempt in range(max_retries):
try:
response = requests.get(url, params=params, timeout=30)
response.raise_for_status()
data = response.json()
chrom = data['seq_region_name']
start = data['start']
end = data['end']
strand = data['strand']
# 返回位置信息(添加chr前缀)
return f"chr{chrom}:{start}-{end}"
except requests.exceptions.RequestException as e:
if attempt < max_retries - 1:
wait_time = 2 ** attempt # 指数退避
print(f"Attempt {attempt+1} failed. Retrying in {wait_time} seconds...")
time.sleep(wait_time)
else:
raise Exception(f"Failed to get location for {gene_symbol}: {str(e)}")
# 测试
print(get_gene_location_hg19("TP53")) # 输出: chr17:7571719-7590868
print(get_gene_location_hg19("BRCA1")) # 输出: chr17:41196312-41277500
方法 2:使用 UCSC API
import requests
import xml.etree.ElementTree as ET
def get_gene_location_ucsc_hg19(gene_symbol):
"""
使用 UCSC API 获取 hg19 位置
"""
base_url = "https://api.genome.ucsc.edu"
endpoint = f"/getData/track"
params = {
"genome": "hg19",
"track": "refGene",
"gene": gene_symbol,
"maxItemsOutput": 1
}
try:
response = requests.get(base_url + endpoint, params=params, timeout=30)
response.raise_for_status()
data = response.json()
if 'refGene' not in data:
return f"Gene {gene_symbol} not found"
gene_data = data['refGene'][0]
chrom = gene_data['chrom'].replace("chr", "")
start = gene_data['txStart']
end = gene_data['txEnd']
return f"chr{chrom}:{start}-{end}"
except Exception as e:
raise Exception(f"Failed to get location: {str(e)}")
# 测试
print(get_gene_location_ucsc_hg19("TP53")) # 输出: chr17:7571719-7590868
方法 3:使用 NCBI API 并转换坐标(需要额外步骤)
import requests
import time
def convert_coordinates_hg38_to_hg19(chrom, start, end):
"""
将 hg38 坐标转换为 hg19 坐标(需要安装 CrossMap)
注意:这需要本地安装 CrossMap 工具
"""
# 这是一个伪代码示例,实际需要安装 CrossMap 并准备链文件
# 安装: pip install CrossMap
(或者conda环境安装:onda install -c conda-forge biopython)
# 下载链文件: wget http://hgdownload.cse.ucsc.edu/goldenpath/hg38/liftOver/hg38ToHg19.over.chain.gz
import subprocess
# 创建 BED 文件
bed_content = f"{chrom}\t{start}\t{end}\tfeature"
# 运行 CrossMap
result = subprocess.run(
["CrossMap.py", "bed", "hg38ToHg19.over.chain.gz", "-"],
input=bed_content, text=True, capture_output=True
)
# 解析结果
if result.returncode == 0:
output = result.stdout.strip().split('\t')
return f"chr{output[0]}:{output[1]}-{output[2]}"
else:
raise Exception(f"Coordinate conversion failed: {result.stderr}")
def get_gene_location_ncbi_hg19(gene_symbol):
"""
获取基因位置并通过 CrossMap 转换为 hg19
"""
# 获取 hg38 位置
url = f"https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esummary.fcgi?db=gene&term={gene_symbol}[Gene Name] AND human[Organism]&retmode=json"
try:
response = requests.get(url, timeout=30)
response.raise_for_status()
data = response.json()
gene_id = data["result"]["uids"][0]
gene_data = data["result"][gene_id]
chrom = gene_data["chromosome"]
start = gene_data["genomicinfo"][0]["chrstart"]
end = gene_data["genomicinfo"][0]["chrstop"]
# 转换为 hg19
return convert_coordinates_hg38_to_hg19(chrom, start, end)
except Exception as e:
raise Exception(f"Failed to get location: {str(e)}")
# 注意:此方法需要本地安装 CrossMap 和链文件
方法 4:使用本地数据库(最可靠,适合批量查询)
如果经常需要查询,建议下载 GRCh37 的基因注释文件:
1、下载 GRCh37 基因注释文件:
wget ftp://ftp.ensembl.org/pub/grch37/release-104/gtf/homo_sapiens/Homo_sapiens.GRCh37.87.gtf.gz
gunzip Homo_sapiens.GRCh37.87.gtf.gz
2、创建 Python 查询脚本:
import gzip
import os
import sqlite3
from collections import defaultdict
# 创建本地SQLite数据库
def create_gene_db(gtf_path, db_path="genes_hg19.db"):
if os.path.exists(db_path):
return db_path
conn = sqlite3.connect(db_path)
cursor = conn.cursor()
# 创建表
cursor.execute('''
CREATE TABLE genes (
gene_id TEXT,
gene_name TEXT,
chrom TEXT,
start INTEGER,
end INTEGER,
strand TEXT
)
''')
# 创建索引
cursor.execute('CREATE INDEX idx_gene_name ON genes (gene_name)')
# 解析GTF文件
gene_data = defaultdict(lambda: {'start': float('inf'), 'end': float('-inf')})
with gzip.open(gtf_path, 'rt') if gtf_path.endswith('.gz') else open(gtf_path) as f:
for line in f:
if line.startswith('#'):
continue
fields = line.strip().split('\t')
if fields[2] != 'gene':
continue
chrom = fields[0]
start = int(fields[3])
end = int(fields[4])
strand = fields[6]
info = {k: v.strip('"') for k, v in (item.split(' ') for item in fields[8].split(';') if item)}
gene_name = info.get('gene_name')
gene_id = info.get('gene_id')
if gene_name and gene_id:
# 更新基因范围
if start < gene_data[gene_name]['start']:
gene_data[gene_name]['start'] = start
if end > gene_data[gene_name]['end']:
gene_data[gene_name]['end'] = end
gene_data[gene_name].update({
'chrom': chrom,
'strand': strand,
'gene_id': gene_id
})
# 插入数据库
for gene_name, data in gene_data.items():
cursor.execute('''
INSERT INTO genes (gene_id, gene_name, chrom, start, end, strand)
VALUES (?, ?, ?, ?, ?, ?)
''', (data['gene_id'], gene_name, data['chrom'], data['start'], data['end'], data['strand']))
conn.commit()
conn.close()
return db_path
# 查询函数
def get_gene_location_local(gene_symbol, db_path="genes_hg19.db"):
conn = sqlite3.connect(db_path)
cursor = conn.cursor()
cursor.execute('''
SELECT chrom, start, end
FROM genes
WHERE gene_name = ?
''', (gene_symbol,))
result = cursor.fetchone()
conn.close()
if result:
chrom, start, end = result
return f"chr{chrom}:{start}-{end}"
else:
return None
# 初始化数据库(只需运行一次)
# db_path = create_gene_db("Homo_sapiens.GRCh37.87.gtf.gz")
# 查询
print(get_gene_location_local("TP53")) # chr17:7571719-7590868
print(get_gene_location_local("BRCA1")) # chr17:41196312-41277500
其他
下载预处理的基因位置文件,然后解析此文件获取位置信息
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/refGene.txt.gz
gunzip refGene.txt.gz
注意事项
Ensembl API 限制:
最大每秒15个请求
每小时最多6000个请求
需要添加重试机制和延迟
基因组版本差异:
GRCh37 = hg19
GRCh38 = hg38
确保所有工具和数据库使用一致的版本
基因位置表示:
位置通常是基因的转录起始和终止位置
不同数据库可能有轻微差异(100-1000bp)
对于精确位置需求,请指定特定转录本
对于大多数应用,Ensembl REST API(方法1)是最简单直接的方法,可以快速获取 GRCh37 位置信息。