文章目录
- 1.前言
- 2.CellNet
- 2.1CellNet简介
- 2.2CellNet结果
- 3.PACNet
- 3.1安装R包与加载R包
- 3.2加载数据
- 3.3开始训练和分类
- 3.4可视化分类过程
- 3.5可视化分类结果
- 4.细胞命运分类和免疫浸润比较
1.前言
今天冲浪看到一个细胞分类性能评估的R包——PACNet,它与转录组分析方法、计算预处理方法和预处理方法产生的基因可用性无关,因此可以对细胞命运工程方案的性能进行交叉研究比较,这个是新包。

其次还有孪生子弟旧R包,已经停止更新了:CellNet

先讲一下CellNet,因为新包的参考数据集也是共用的,但使用的话我们还是用PACNet哈
2.CellNet
2.1CellNet简介
CellNet是一个基于网络生物学的计算平台,用于评估细胞工程的保真度,并生成用于改进细胞衍生的假设。CellNet基于细胞类型特异性基因调控网络(GRN)的重建,有16种小鼠和16种人类细胞和组织类型的公开RNA-Seq数据进行重建。
简单过一下CellNet,目前,有两种方法可以运行CellNet获取RNA-Seq数据。作者提供了云平台和本地运行两种方式,因为亚马逊云要氪金,本着白嫖的意思就本地跑一跑了
- Cutadapt
- Salmon
- GNU Parallel
CellNet的核心是随机森林分类器。这是对细胞命运实验结果进行分类的算法。要使用 CellNet 分析自己的表达数据,需要一个经过训练的 CellNet 分类器对象,我们将其称为 cnProc(CellNet 处理器)。
第一步需要先构建cnProc对象,可以自己去构建,相关代码:构建cnProc,优势就是可以增加自己需要研究细胞类型,或者研究人鼠外其他物种。
可以使用作者整理好了的rdata,在github中下载即可:

第二第三步就是RNA数据(要SRA文件)和索引了
作者也提供了:

2.2CellNet结果
- 分类热图:在训练数据(行)中显示每个样本(列)对每个细胞和组织类型的分类分数:
pdf(file='hmclass_example.pdf', width=7, height=5)
cn_HmClass(cnRes)
dev.off()
这里可以看到iPSC在胚胎干细胞中分数更高,其次是Day0的几个在成纤维细胞中分数更高。

- Gene Regulatory Network 状态栏图:一种更灵敏的测量,用于测量特定细胞类型的 GRN 在实验数据中建立的程度
fname<-'grnstats_esc_example.pdf'
bOrder<-c("fibroblast_train", unique(as.vector(stQuery$description1)), "esc_train")
cn_barplot_grnSing(cnRes,cnProc,"esc", c("fibroblast","esc"), bOrder, sidCol="sra_id")
ggplot2::ggsave(fname, width=5.5, height=5)
dev.off()
这里关于基因调控网络状态的,如果说热图是一个计算分数绝对值的匹配,那这里就是对调控网络状态,一个动态的匹配

- Network Influence Score Box and Whisker Plot:可以更好地调节的转录因子的建议,按其潜在影响进行排序
rownames(stQuery)<-as.vector(stQuery$sra_id)
tfScores<-cn_nis_all(cnRes, cnProc, "esc")
fname<-'nis_esc_example_Day0.pdf'
plot_nis(tfScores, "esc", stQuery, "Day0", dLevel="description1", limitTo=0)
ggplot2::ggsave(fname, width=4, height=12)
dev.off()
这个就调控影响分数的排序

3.PACNet
流程和输入文件是与CellNet一样的,直接跳过开始demo
3.1安装R包与加载R包
install.packages("devtools")
library(devtools)
install_github("pcahan1/CellNet", ref="master")
install_github("pcahan1/cancerCellNet@v0.1.1", ref="master")
source("pacnet_utils.R")
library(CellNet)
library(cancerCellNet)
library(plyr)
library(ggplot2)
library(RColorBrewer)
library(pheatmap)
library(plotly)
library(igraph)
3.2加载数据
这里就需要表达矩阵和元数据列表。
表达矩阵应将基因符号作为行名,将样本名称作为列名。示例元数据表应将示例名称作为行名,将示例要素作为列名。表达式矩阵的列名必须与元数据表的行名匹配。为了使分类器训练可靠,每种训练类型至少应有 60 个独立rep。
元数据列表的title格式:

加载数据
expTrain <- utils_loadObject("Hs_expTrain_Jun-20-2017.rda")
stTrain <- utils_loadObject("Hs_stTrain_Jun-20-2017.rda")
加载工程参考数据和查询数据
liverRefExpDat <- utils_loadObject("liver_engineeredRef_normalized_expDat_all.rda")
liverRefSampTab <- utils_loadObject("liver_engineeredRef_sampTab_all.rda")
queryExpDat <- read.csv("example_data/example_counts_matrix.csv", row.names=1)
querySampTab <- read.csv("example_data/example_sample_metadata_table.csv")
rownames(querySampTab) <- querySampTab$sample_name
study_name <- "liver_example"
识别交叉基因:
iGenes <- intersect(rownames(expTrain), rownames(liverRefExpDat))
iGenes <- intersect(iGenes, rownames(queryExpDat))
# Subset training expression matrix based on iGenes
expTrain <- expTrain[iGenes,]
3.3开始训练和分类
将数据拆分为训练集和验证集:
set.seed(99) # Setting a seed for the random number generator allows us to reproduce the same split in the future
stList <- splitCommon_proportion(sampTab = stTrain, proportion = 0.66, dLevel = "description1") # Use 2/3 of training data for training and 1/3 for validation
stTrainSubset <- stList$trainingSet
expTrainSubset <- expTrain[,rownames(stTrainSubset)]
#See number of samples of each unique type in description1 in training subset
table(stTrainSubset$description1)
stValSubset <- stList$validationSet
expValSubset <- expTrain[,rownames(stValSubset)]
#See number of samples of each unique type in description1 in validation subset
table(stValSubset$description1)
训练随机森林分类器,需要 3-10 分钟,具体取决于内存可用性:
system.time(my_classifier <- broadClass_train(stTrain = stTrainSubset,
expTrain = expTrainSubset,
colName_cat = "description1",
colName_samp = "sra_id",
nRand = 70,
nTopGenes = 100,
nTopGenePairs = 100,
nTrees = 2000,
stratify=TRUE,
sampsize=25, # Must be less than the smallest number in table(stTrainSubset$description1)
quickPairs=TRUE)) # Increasing the number of top genes and top gene pairs increases the resolution of the classifier but increases the computing time
save(my_classifier, file="example_outputs/cellnet_classifier_100topGenes_100genePairs.rda")
3.4可视化分类过程
- 分类热图:
stValSubsetOrdered <- stValSubset[order(stValSubset$description1), ] #order samples by classification name
expValSubset <- expValSubset[,rownames(stValSubsetOrdered)]
cnProc <- my_classifier$cnProc #select the cnProc from the earlier class training
classMatrix <- broadClass_predict(cnProc, expValSubset, nrand = 60)
stValRand <- addRandToSampTab(classMatrix, stValSubsetOrdered, desc="description1", id="sra_id")
grps <- as.vector(stValRand$description1)
names(grps)<-rownames(stValRand)
# Create validation heatmap
png(file="classification_validation_hm.png", height=6, width=10, units="in", res=300)
ccn_hmClass(classMatrix, grps=grps, fontsize_row=10)
dev.off()

- 验证精度-召回率曲线:
assessmentDat <- ccn_classAssess(classMatrix, stValRand, classLevels="description1", dLevelSID="sra_id")
png(file="example_outputs/classifier_assessment_PR.png", height=8, width=10, units="in", res=300)
plot_class_PRs(assessmentDat)
dev.off()

- 基因对验证:
genePairs <- cnProc$xpairs
# Get gene to gene comparison of each gene pair in the expression table
expTransform <- query_transform(expTrainSubset, genePairs)
avgGenePair_train <- avgGeneCat(expDat = expTransform, sampTab = stTrainSubset,
dLevel = "description1", sampID = "sra_id")
genePairs_val <- query_transform(expValSubset, genePairs)
geneCompareMatrix <- makeGeneCompareTab(queryExpTab = genePairs_val,
avgGeneTab = avgGenePair_train, geneSamples = genePairs)
val_grps <- stValSubset[,"description1"]
val_grps <- c(val_grps, colnames(avgGenePair_train))
names(val_grps) <- c(rownames(stValSubset), colnames(avgGenePair_train))
png(file="example_outputs/validation_gene-pair_comparison.png", width=10, height=80, units="in", res=300)
plotGeneComparison(geneCompareMatrix, grps = val_grps, fontsize_row = 6)
dev.off()
该图较大,主要就是基因对的分数热图,这个就是xpairs图

创建并保存xpairs_list对象,用于 grn 重建和训练规范化参数:
xpairs_list <- vector("list", 14)
for (pair in rownames(avgGenePair_train)) {
for (j in 1:ncol(avgGenePair_train)) {
if (avgGenePair_train[pair,j] >= 0.5) {
if (is.null(xpairs_list[[j]])) {
xpairs_list[[j]] <- c(pair)
} else {
xpairs_list[[j]] <- c(xpairs_list[[j]], pair)
}
}
}
}
xpair_names <- colnames(avgGenePair_train)
xpair_names <- sub(pattern="_Avg", replacement="", x=xpair_names)
names(xpairs_list) <- xpair_names
for (type in names(xpairs_list)) {
names(xpairs_list[[type]]) <- xpairs_list[[type]]
}
save(xpairs_list, file="example_outputs/Hs_xpairs_list.rda")
3.5可视化分类结果
对训练集样本进行分类
classMatrixLiverRef <- broadClass_predict(cnProc = cnProc, expDat = liverRefExpDat, nrand = 10)
grp_names1 <- c(as.character(liverRefSampTab$description1), rep("random", 10))
names(grp_names1) <- c(as.character(rownames(liverRefSampTab)), paste0("rand_", c(1:10)))
# Re-order classMatrixQuery to match order of rows in querySampTab
classMatrixLiverRef <- classMatrixLiverRef[,names(grp_names1)]
png(file="example_outputs/heatmapLiverRef.png", height=12, width=9, units="in", res=300)
heatmapRef(classMatrixLiverRef, liverRefSampTab) # This function can be found in pacnet_utils.R
dev.off()
# Alternatively, for an interactive plotly version:
heatmapPlotlyRef(classMatrixLiverRef, liverRefSampTab)

对测试集进行分类:
queryExpDat <- log(1+queryExpDat)
classMatrixQuery <- broadClass_predict(cnProc = cnProc, expDat = queryExpDat, nrand = 3)
grp_names <- c(as.character(querySampTab$description1), rep("random", 3))
names(grp_names) <- c(as.character(rownames(querySampTab)), paste0("rand_", c(1:3)))
# Re-order classMatrixQuery to match order of rows in querySampTab
classMatrixQuery <- classMatrixQuery[,names(grp_names)]
save(classMatrixQuery, file="example_outputs/example_classificationMatrix.rda")
png(file="example_outputs/query_classification_heatmap.png", height=4, width=8, units="in", res=300)
# This function can be found in pacnet_utils.R
acn_queryClassHm(classMatrixQuery, main = paste0("Classification Heatmap, ", study_name),
grps = grp_names,
fontsize_row=10, fontsize_col = 10, isBig = FALSE)
dev.off()

计算调控因子得分
## 准备用于网络影响得分计算的 GRN 和表达式数据
## 基于交叉基因的子集和对象:grnAll,trainNormParam
grnAll <- utils_loadObject("liver_grnAll.rda")
trainNormParam <- utils_loadObject("liver_trainNormParam.rda")
# These two functions can be found in pacnet_utils.R
grnAll <- subsetGRNall(grnAll, iGenes)
trainNormParam <- subsetTrainNormParam(trainNormParam, grnAll, iGenes)
queryExpDat_ranked <- logRank(queryExpDat, base = 0)
queryExpDat_ranked <- as.data.frame(queryExpDat_ranked)
## 计算转录调节因子的计算网络影响评分 (NIS)
network_cell_type <- "liver"
target_cell_type <- "liver"
system.time(TF_scores <- pacnet_nis(expDat = queryExpDat_ranked, stQuery=querySampTab, iGenes=iGenes,
grnAll = grnAll, trainNorm = trainNormParam,
subnet = network_cell_type, ctt=target_cell_type,
colname_sid="sample_name", relaWeight=0))
save(TF_scores, file="example_outputs/my_study_TF_scores.rda")
## 选择得分最高的 25 个 TF 进行绘图:
TFsums <- rowSums(abs(TF_scores))
ordered_TFsums <- TFsums[order(TFsums, decreasing = TRUE)]
if(length(TFsums) > 25) {
top_display_TFs <- names(ordered_TFsums)[1:25]
} else {
top_display_TFs <- names(ordered_TFsums)
}
TF_scores <- TF_scores[top_display_TFs,]
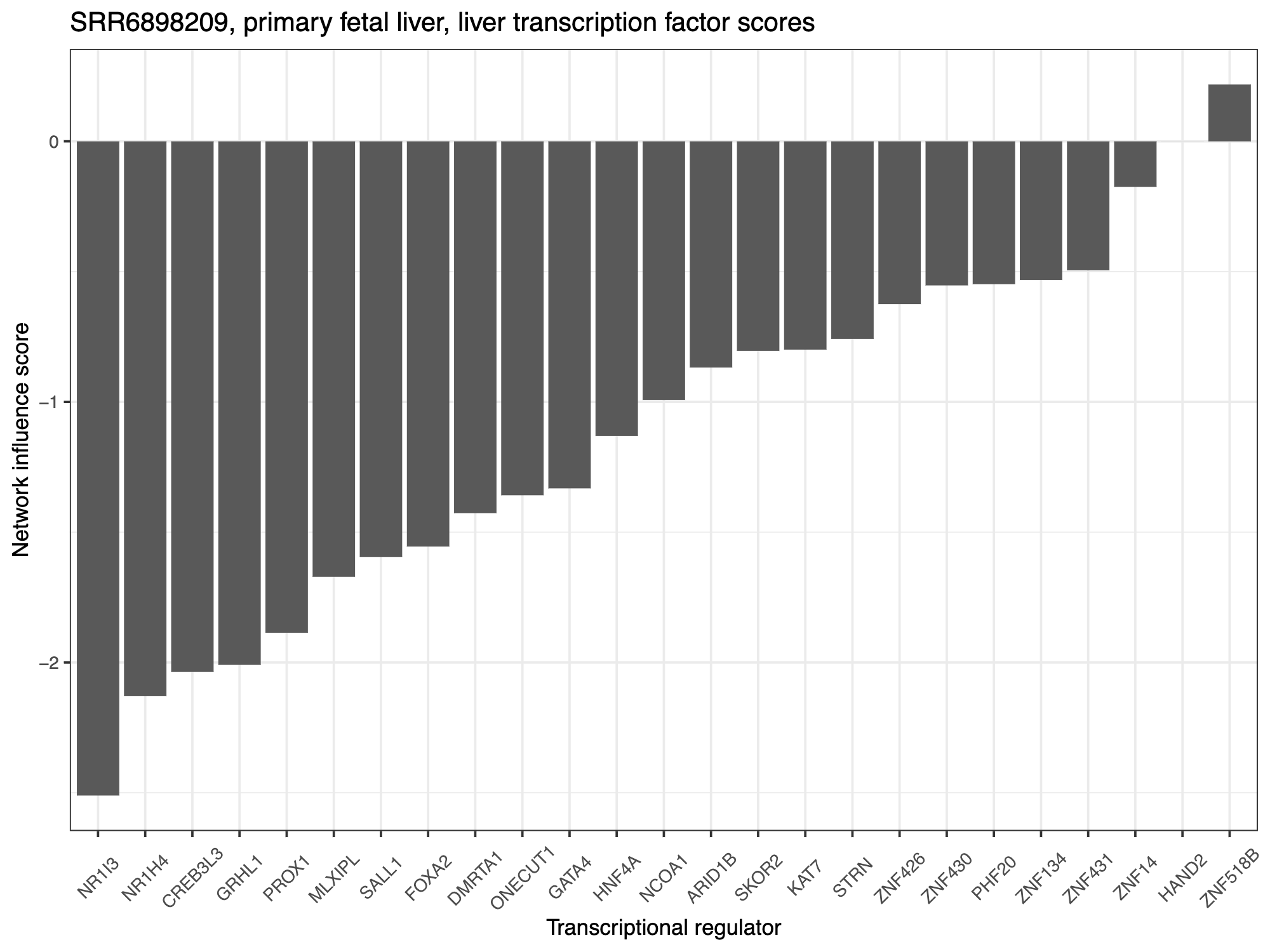
## 绘制 TF 分数:
sample_names <- rownames(querySampTab)
pdf(file="example_outputs/my_study_TF_scores_my_cell_type.pdf", height=6, width=8)
for(sample in sample_names) {
descript <- querySampTab$description1[which(rownames(querySampTab) == sample)]
plot_df <- data.frame("TFs" = rownames(TF_scores),
"Scores" = as.vector(TF_scores[,sample]))
sample_TFplot <- ggplot(plot_df, aes(x = reorder(TFs,Scores,mean) , y = Scores)) +
geom_bar(stat="identity") + #aes(fill = medVal)) +
theme_bw() +
ggtitle(paste0(sample, ", ", descript, ", ", target_cell_type, " transcription factor scores")) +
ylab("Network influence score") + xlab("Transcriptional regulator") +
theme(legend.position = "none", axis.text = element_text(size = 8)) +
theme(text = element_text(size=10),
legend.position="none",
axis.text.x = element_text(angle = 45, vjust=0.5))
print(sample_TFplot)
}
dev.off()
阴性 TF 评分表明给定的 TF 应该上调以获得与靶细胞类型更相似的身份。正 TF 评分表明给定的 TF 应下调以获得与靶细胞类型更相似的身份。
4.细胞命运分类和免疫浸润比较
- 免疫浸润评估主要是指在特定组织(如肿瘤组织)中,不同类型的免疫细胞的存在与活性的分析。这涉及到分析如何及在什么程度上各种免疫细胞(例如
T细胞、B细胞、巨噬细胞等)参与到组织的免疫应答中。免疫浸润的水平可以作为疾病预后的一个重要指标,特别是在癌症研究中,高水平的免疫浸润通常与较好的预后相关。 - 拿常见的CIBERSORT来看,这是用于从复杂的组织表达数据中,通过特征矩阵例如LM22估计细胞组成的相对丰度的免疫浸润方法。
- PACNet是用于分析特别是在癌症研究中常见的多组分样本(如肿瘤微环境中的细胞)。它提供了一种网络方法,通过整合表达数据和先验分子网络信息,来预测样本中细胞类型的丰度。
- 各有侧重,CIBERSORT 更专注于从复杂组织样本中准确估计免疫细胞的丰度,而 PACNet 则提供了一种网络分析方法,不仅可以估计细胞丰度,还可以探究细胞之间的相互作用和网络结构。
对于做分化、做干细胞、做肿瘤分型等来说,这真是一大利器,埋下伏笔,下期更新单细胞的~





![[论文笔记]Root Mean Square Layer Normalization](https://img-blog.csdnimg.cn/img_convert/ebea04d0e75943d4b3eeb5b2449b92db.png)