目录
基本概念
基本原理
下游分析
基本概念
WGCNA其译为加权基因共表达网络分析。该分析方法旨在寻找协同表达的基因模块(module),并探索基因网络与关注的表型之间的关联关系,以及网络中的核心基因。
适用于复杂的数据模式(推荐5组(或者15个样品)以上的数据)。一般可应用的研究方向有:不同器官或组织类型发育调控、同一组织不同发育调控、非生物胁迫不同时间点应答。
基本原理
从方法上来讲,WGCNA分为表达量聚类分析和表型关联两部分,主要包括基因之间相关系数计算、基因模块的确定、共表达网络、模块与性状关联四个步骤。
第一步计算任意两个基因之间的相关系数(Person Coefficient)。为了衡量两个基因是否具有相似表达模式,一般需要设置阈值来筛选,高于阈值的则认为是相似的。但是这样如果将阈值设为0.8,那么很难说明0.8和0.79两个是有显著差别的。因此,WGCNA分析时采用相关系数加权值,即对基因相关系数取N次幂,使得网络中的基因之间的连接服从无尺度网络分布(scale-freenetworks),这种算法更具生物学意义。
第二步通过基因之间的相关系数构建分层聚类树,聚类树的不同分支代表不同的基因模块,不同颜色代表不同的模块。基于基因的加权相关系数,将基因按照表达模式进行分类,将模式相似的基因归为一个模块。这样就可以将几万个基因通过基因表达模式被分成了几十个模块,是一个提取归纳信息的过程。
下游分析
得到模块之后的分析有:
1.模块的功能富集(见参考来源文章)
2.模块与性状之间的相关性(见参考来源文章)
3.模块与样本间的相关系数
挖掘模块的关键信息:
1.找到模块的核心基因
2.利用关系预测基因功能
本研究使用 R 语言的 WGCNA 包(Langfelder and Horvath, 2008)进行加权基因
共表达网络分析,详细的步骤如下:
(
1
)将
49
个样本的
18,964
个基因的表达值组合成一个
TPM
表达值矩阵(使用的是对转录组数据进行标准化TPM后),并将 其作为 WGCNA
的输入文件进行网络构建;
(2)使用函数“
goodSamplesGenes
”检测并删除具有过多缺失值的基因和样本;
(3)使用函数“
pickSoftThreshold
”在
1
到
30
之间确定最合适的软阈值,使构
建的共表达网络更符合无标度网络的特征;
(4)使用最合适的软阈值
18
,通过函数“
adjacency
”计算有向网络(
signed
WGCNA network
)的邻接矩阵(
adjacency matrix
);
(
5
)以邻接矩阵作为输入,通过函数“
TOMsimilarity
”计算拓扑重叠矩阵
(
topological overlap matrix
,
TOM
);
(6)使用拓扑重叠矩阵作为输入,利用动态树剪切法(
dynamic cut tree algorithm
),
通过函数“
cutreeDynamic
”进行共表达模块的探测。并且在这个最初的网络构建中,
最小模块大小被设置为
30
个基因;
(7)使用函数“
moduleEigengenes
”将距离小于
0.1
的模块进行合并。
(8)每一个模块的特征基因(
module eigengene
,
ME)通过主成分分析进行计算。
并使用基因表达矩阵的第一主成分作为模块特征基因。
通过以上步骤,基因表达水平高度正相关的基因被分配到了相同的共表达模块。
其中未分配到任何模块的基因被保存进入 ME0 模块中,实际上它并不是一个真正的
模块。
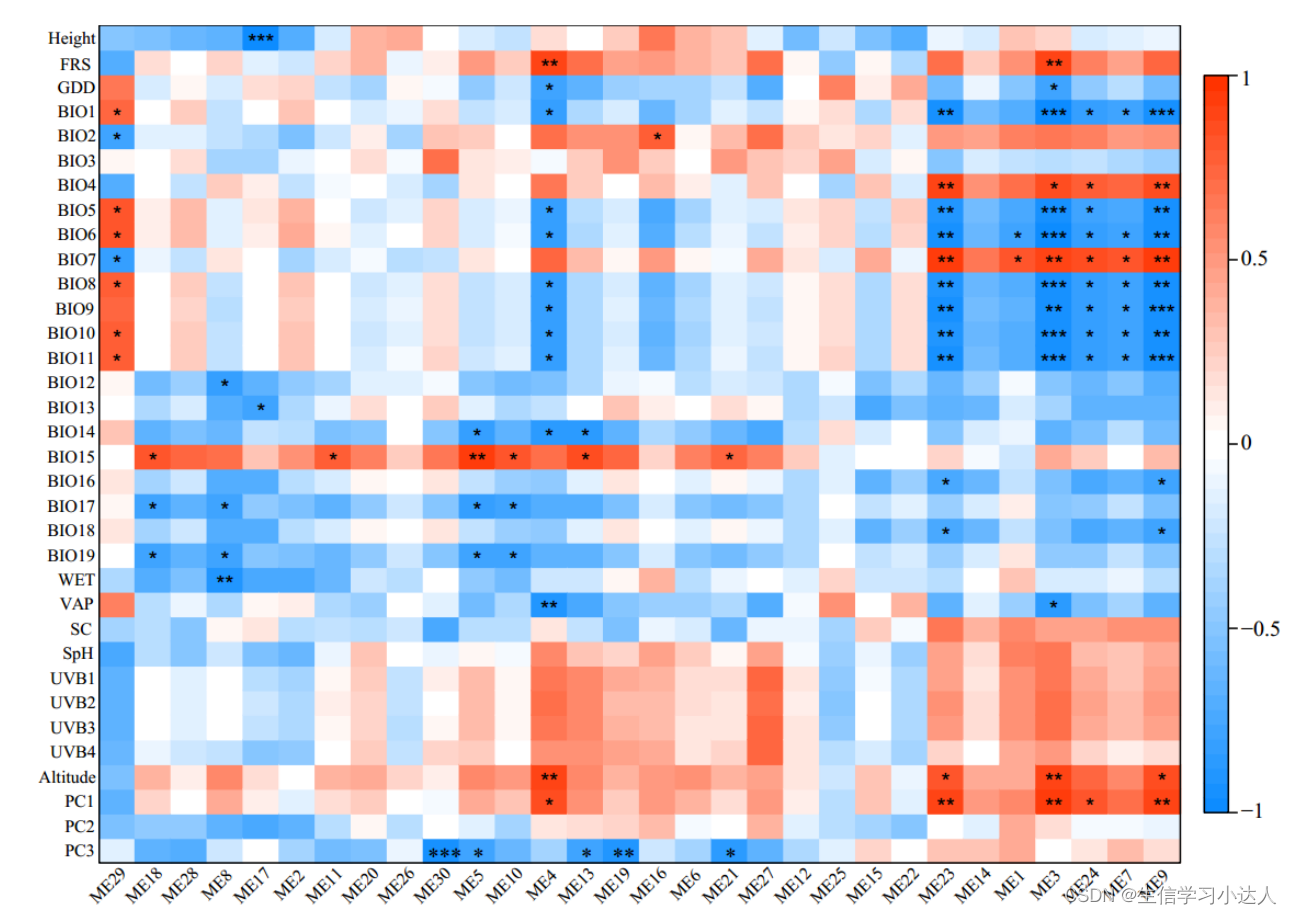
参考来源:
WGCNA分析
云南松遗传变异格局及适应性分化的遗传基础研究_孙彦强
模块和性状关联并识别模块的核心基因









![[lesson33]C++中的字符串类](https://img-blog.csdnimg.cn/direct/8ec2075e3c5a4a799ddf7bf4fc606012.png#pic_center)