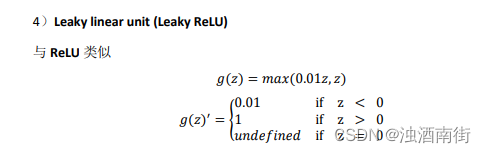上次,我们对测序数据去了人源序列。接下来我们就要对去人源的reads进行微生物物种注释。
我们选择karken2和bracken。
首先是建立karken2的数据库,有多种方法。
方法一:kraken2-build --standard --threads 4 --db ./standardDB #时间太慢
方法二:直接下载官方提供的数据库(包括kraken2及bracken),解压即可
wget https://genome-idx.s3.amazonaws.com/kraken/k2_pluspf_20210517.tar.gz
wget https://genome-idx.s3.amazonaws.com/kraken/k2_pluspf_8gb_20210517.tar.gz
wget https://genome-idx.s3.amazonaws.com/kraken/k2_standard_20210517.tar.gz #时间太慢
# 方法3:手动构建数据库 #时间太慢
## step1 从 NCBI 下载分类文件
kraken2-build --download-taxonomy --db ./K2db
## step2 下载序列数据
kraken2-build --download-library UniVec_Core --threads 15 --db ./K2db
kraken2-build --download-library UniVec --threads 4 --15 ./K2db
## step3 构建数据库
kraken2-build --build --threads 4 --db ./K2db
前三种时间都比较慢,所以我不用。
第四种方法为 在windows环境下登录官网https://benlangmead.github.io/aws-indexes/k2下载自己想要的数据库,官网中有诸多类型的数据库。
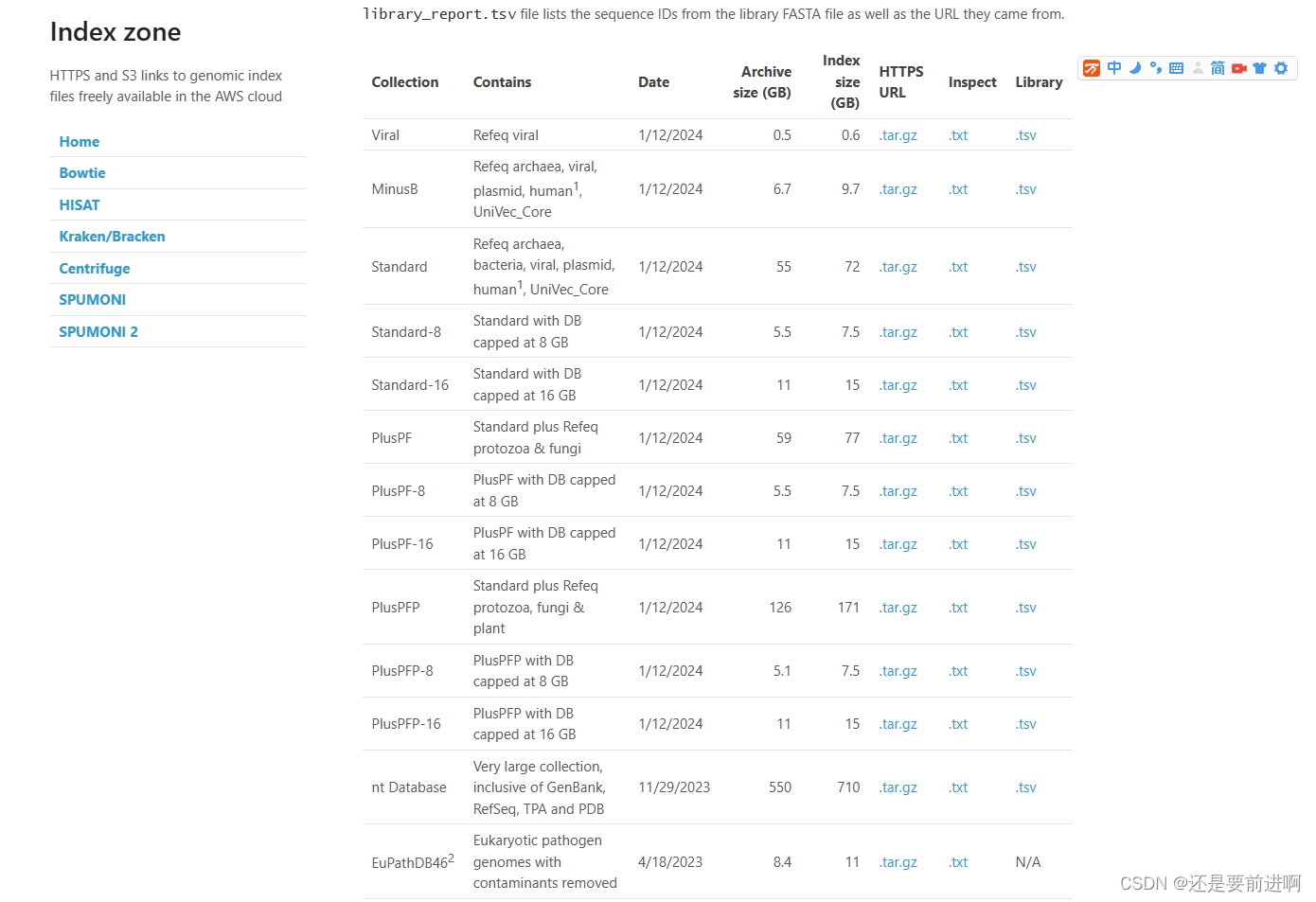
官网里面有很多已经做好的数据库。我们可以选择合适的使用。
我这里先下载 standard-8 数据库
#解压缩,kracken和bracken的数据就都好了。
tar -zxvf k2_standard_08gb_20231009.tar.gz
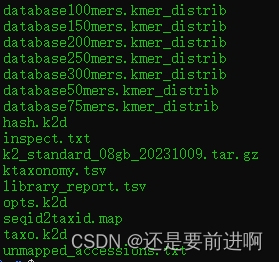
解压缩出来的是这些文件。
接下来就可以跑数据了。
kraken2 --threads 15 --paired --db /mnt/h/db/kraken2.db --report A1.kreport --output A1.kraken read_hont_removed.1.fq.gz read_hont_removed.2.fq.gz
-
kraken2: 运行 Kraken 2 软件。 -
--threads 15: 指定使用的线程数为 15,即并行处理的线程数。 -
--paired: 表示输入的是 paired-end 测序数据。 -
--db /mnt/h/db/kraken2.db: 指定 Kraken 2 数据库的路径,即要用于比对和分类的数据库。 -
--report A1.kreport: 指定输出报告文件的名称为 A1.kreport,该报告包含了分类和注释的结果信息。 -
--output A1.kraken: 指定输出文件的名称为 A1.kraken,该文件包含了每个 reads 的分类结果。 -
read_hont_removed.1.fq.gz read_hont_removed.2.fq.gz: 输入的 paired-end 测序数据文件,分别是第一端和第二端的 fastq 文件。
接下来就是bracken.
bracken -d /mnt/h/db/kraken2.db -i A1.kreport -o A1.bracken.S -w A1.bracken.S.kreport -l S -t 15
-
bracken: 运行 Bracken 软件。 -
-d /mnt/h/db/kraken2.db: 指定 Kraken 2 数据库的路径,即要用于物种丰度估计的数据库。 -
-i A1.kreport: 指定输入的 Kraken 2 分类报告文件,即 A1.kreport。 -
-o A1.bracken.S: 指定输出文件的名称为 A1.bracken.S,该文件包含了物种丰度的估计结果。 -
-w A1.bracken.S.kreport: 指定输出详细报告文件的名称为 A1.bracken.S.kreport,该文件包含了对每个分类水平的物种丰度估计结果。 -
-l S: 指定要进行物种丰度估计的分类水平为 species level(物种水平)。 -
-t 15: 指定使用的线程数为 15,即并行处理的线程数。
这就是karken2和bracken最基础的应用了,想要结果更好,还需向深处探索。
我推荐文章《Metagenome analysis using the Kraken software suite》
《Kraken: ultrafast metagenomic sequence classification using exact alignments》



![[CSS]中块级格式化上下文(BFC)](https://img-blog.csdnimg.cn/direct/a8409481a47948bf99d4c5fe835d1509.png)