miRNA测序数据生信分析——第三讲,已知物种的生信分析实例
- miRNA测序数据生信分析——第三讲,已知物种的生信分析实例
- 1. 下载测序数据
- 2. 原始数据质控——软件fastqc
- 3. 注释tRNA和rRNA,使用Rfam数据库——软件blast,Rfam_statistics.py脚本
- 4. 注释miRNA,包括种类,序列及定量,靶基因和绘图
- 4.1 鉴定,使用miRBase数据库——软件blast
- 4.2 定量和miRNA序列提取——脚本miRBase_sequence.py
- 4.3 miRNA靶基因,使用miRTarBase和miRDB数据库
- 4.3.1 miRTarBase数据库——脚本miRTarBase_Target.py
- 4.3.2 miRDB数据库——脚本miRDB_Target.py
- 4.3.3 整合两个数据库——脚本Total_Target.py
- 4.4 绘制miRNA-靶基因互作图——软件Cytoscape
- 5. 总结
miRNA测序数据生信分析——第三讲,已知物种的生信分析实例
1. 下载测序数据
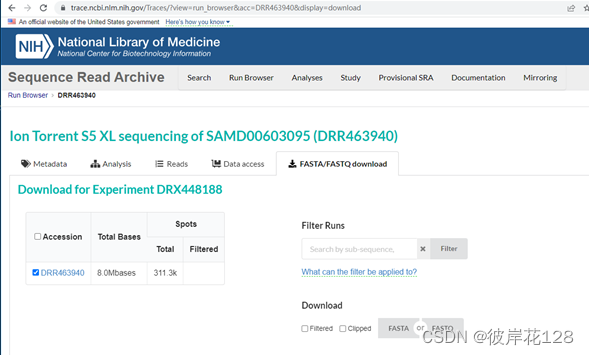
SRA号:DRR463940 单端测序 测序类型:miRNA-seq
点击FASTQ,下载即可。文件DRR463940.fastq
2. 原始数据质控——软件fastqc
cd /home/zhaohuiyao/miRNA_seq/DRR463940/00Rawdata
#质控
/home/zhaohuiyao/Biosoft/general/FastQC/fastqc ./DRR463940.fastq
#Read数目:311289;Read长度分布:8~136bp
#查看质控下的每一个模块,都是可以理解的,判断不修剪
/home/zhaohuiyao/Biosoft/seqkit fq2fa -w 0 ./DRR463940.fastq > ./DRR463940.fasta
3. 注释tRNA和rRNA,使用Rfam数据库——软件blast,Rfam_statistics.py脚本
这里需要的Rfam数据库数据是博文:miRNA测序数据生信分析——第二讲,数据库下载整理,中提到的1.2.2 用于注释ncRNA/sRNA测序中的tRNA和rRNA序列,整理的。
为什么要做这一步呢?
从第二步质控结果Read长度分布:8~136bp,判断虽然是miRNA测序,但是依旧有rRNA和tRNA混入。做这一步,可以看看混入占比。
cd /home/zhaohuiyao/miRNA_seq/DRR463940/01Rfam
#只保留一个比对结果
/home/zhaohuiyao/Biosoft/general/ncbi-blast-2.10.0+/bin/blastn -db /home/zhaohuiyao/Database/Rfam/Rfam -query …/00Rawdata/DRR463940.fasta -out DRR463940_Rfam.annotations -outfmt 6 -evalue 1e-5 -num_alignments 1 -num_threads 36
#统计
python ./Rfam_statistics.py -i ./DRR463940_Rfam.annotations -db1 /home/zhaohuiyao/Database/Rfam/family.txt -db2 /home/zhaohuiyao/Database/Rfam/Rfam.full_region -o ./
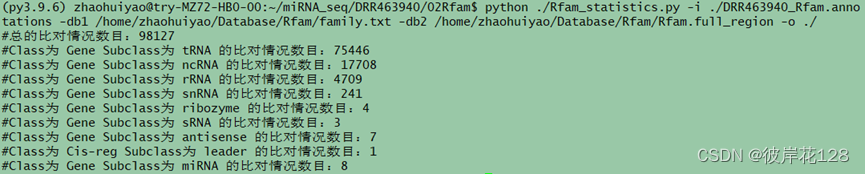
#注意1:这里Subclass为ncRNA指在Rfam数据库中定义了Class但没有定义Subclass的ncRNA。注意2:可以看中重点比对结果出现在tRNA和rRNA,而其他注释类型少。
#结果
#总比对结果数目:98127条(98127/311289=31.52%)
#tRNA比对结果数目:75446条(75446/311289=24.24%)
#rRNA比对结果数目:4709条(4709/311289=1.51%)
4. 注释miRNA,包括种类,序列及定量,靶基因和绘图
测序物种已知,人类Homo sapiens(hsa)。且该物种在后续使用的miRBase、miRDB、miRTarbase数据库中都存在。
4.1 鉴定,使用miRBase数据库——软件blast
cd /home/zhaohuiyao/miRNA_seq/DRR463940/02miRNA/known/
grep “Homo sapiens” /home/zhaohuiyao/Database/miRBase/organisms.txt

#提取miRBase数据库中物种hsa的所有miRNA序列,制作物种特异数据库。
grep -A 1 “hsa” /home/zhaohuiyao/Database/miRBase/mature.fa | grep -v “--” > /home/zhaohuiyao/Database/miRBase/hsa_mature.fa
grep -c “>” /home/zhaohuiyao/Database/miRBase/hsa_mature.fa #2656个miRNA
/home/zhaohuiyao/Biosoft/general/ncbi-blast-2.10.0+/bin/makeblastdb -in /home/zhaohuiyao/Database/miRBase/hsa_mature.fa -dbtype nucl -out /home/zhaohuiyao/Database/miRBase/hsa_mature
#只保留一个比对结果
cd /home/zhaohuiyao/miRNA_seq/DRR463940/02miRNA/known/01miRBase
/home/zhaohuiyao/Biosoft/general/ncbi-blast-2.10.0+/bin/blastn -task blastn-short -db /home/zhaohuiyao/Database/miRBase/hsa_mature -query /home/zhaohuiyao/miRNA_seq/DRR463940/00Rawdata/DRR463940.fasta -out DRR463940_miRBase.annotations -outfmt 6 -evalue 1e-5 -num_alignments 1
#统计
wc -l ./DRR463940_miRBase.annotations #66776条比对结果(66776/311289=21.45%)
cut -f 2 ./DRR463940_miRBase.annotations | sort | uniq | wc -l #367种miRNA
4.2 定量和miRNA序列提取——脚本miRBase_sequence.py
cd /home/zhaohuiyao/miRNA_seq/DRR463940/02miRNA/known/02Sequence_Quantity
python ./miRBase_sequence.py -i …/01miRBase/DRR463940_miRBase.annotations -db /home/zhaohuiyao/Database/miRBase/hsa_mature.fa -o ./

#两个结果文件:
DRR463940_miRBase.annotations.fa和DRR463940_miRBase.annotations.readscount


4.3 miRNA靶基因,使用miRTarBase和miRDB数据库
#三个子目录miRTarBase/、miRDB/和Total/
4.3.1 miRTarBase数据库——脚本miRTarBase_Target.py
cd /home/zhaohuiyao/miRNA_seq/DRR463940/02miRNA/known/03Target/miRTarBase
#确保物种在miRTarBase数据库中
grep “hsa” /home/zhaohuiyao/Database/miRTarBase/miRTarBase.organism

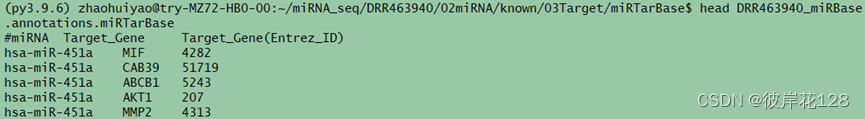
python ./miRTarBase_Target.py -i …/…/02Sequence_Quantity/DRR463940_miRBase.annotations.readscount -db /home/zhaohuiyao/Database/miRTarBase/miRTarBase_MTI.txt -o ./
#结果文件DRR463940_miRBase.annotations.miRTarBase
4.3.2 miRDB数据库——脚本miRDB_Target.py
cd /home/zhaohuiyao/miRNA_seq/DRR463940/02miRNA/known/03Target/miRDB
#确保物种在miRDB数据库中
grep “hsa” /home/zhaohuiyao/Database/miRTarBase/miRDB.organism

python ./miRDB_Target.py -i …/…/02Sequence_Quantity/DRR463940_miRBase.annotations.readscount -db /home/zhaohuiyao/Database/miRDB/miRDB_v6.0_prediction_result.txt.hsa -o ./
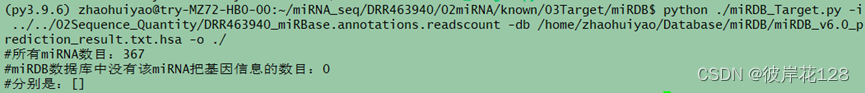
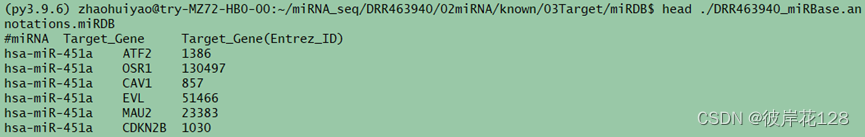
#结果文件DRR463940_miRBase.annotations.miRDB
4.3.3 整合两个数据库——脚本Total_Target.py
#取两个数据库的并集,获得最终miRNA-Gene关系文件
cd /home/zhaohuiyao/miRNA_seq/DRR463940/02miRNA/known/03Target/Total
python ./Total_Target.py -db1 …/miRTarBase/DRR463940_miRBase.annotations.miRTarBase -db2 …/miRDB/DRR463940_miRBase.annotations.miRDB -o ./

#结果文件DRR463940_miRBase.annotations.target

4.4 绘制miRNA-靶基因互作图——软件Cytoscape
因为这个互作关系很庞大,有351413条关系。因此绘制会比较难,我就单独提取了部分互作关系,进行绘图,在Windows下进行。绘图查看另一篇公众号文章:https://mp.weixin.qq.com/s/vbFAre601-9atwah9PMwUw查看
5. 总结
以上就是针对已知物种的miRNA分析。同时满足miRBase、miRTarBase和miRDB三个数据的物种,只有5种。因此针对未知的分析是重要的,而且在你时候的时候,可能会交叉使用。上面步骤中涉及了很多脚本,但都是很简单的文件内容提取比对。