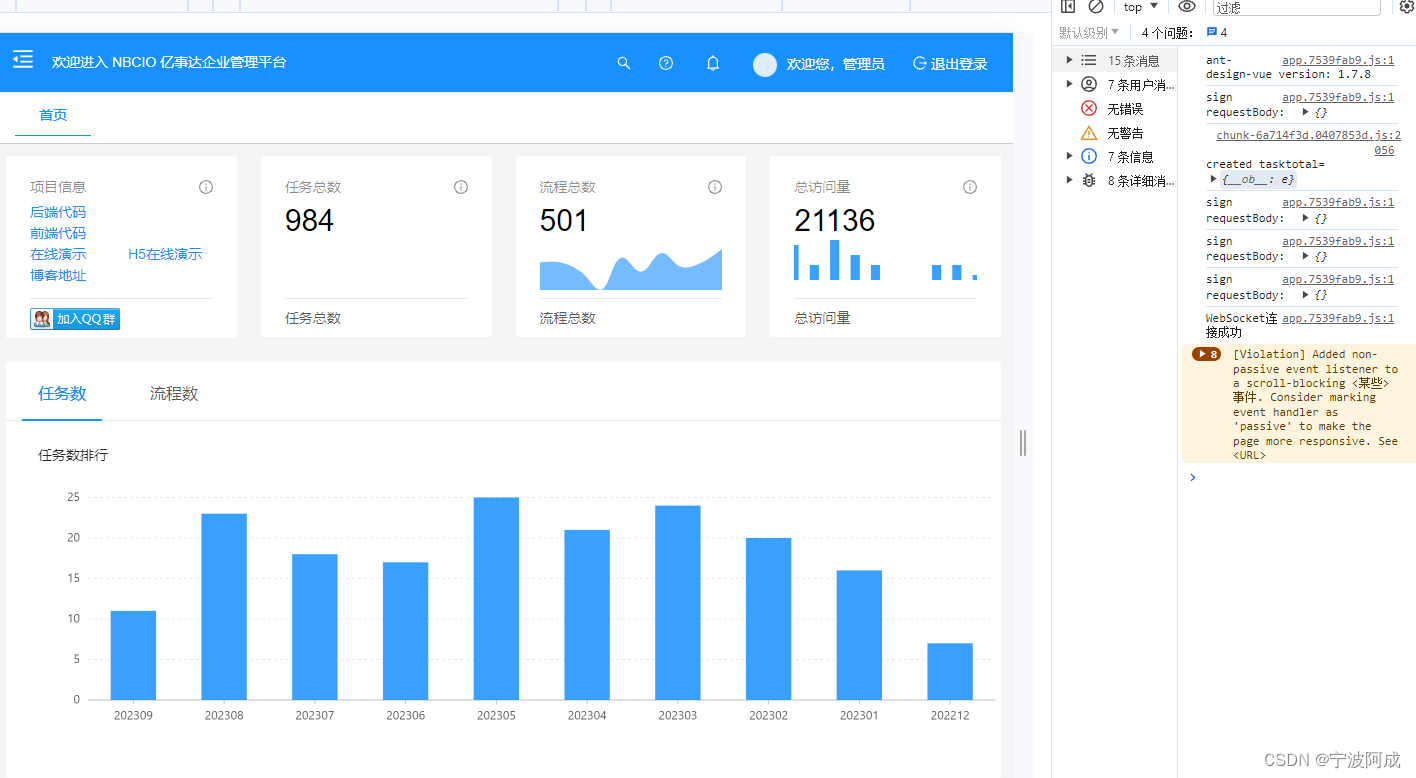
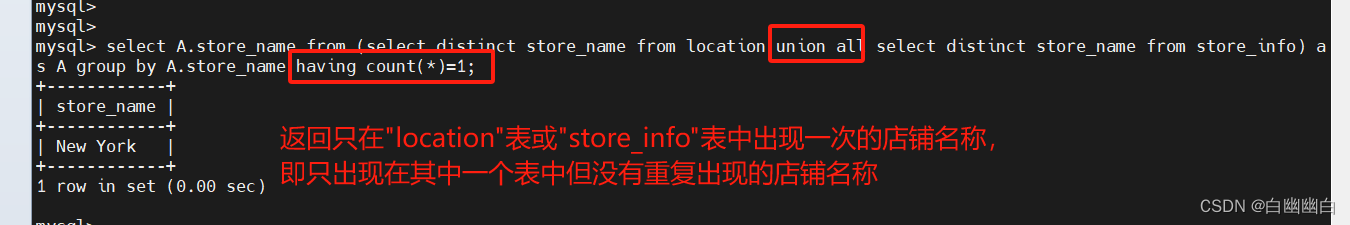
GROMACS Tutorial 1: Lysozyme in Water 中文实战教程
- 前言
- 系统环境
- 特别强调
- 一、预处理阶段
- 1.1 补全原子或残基
- 1.2 删除水分子
- 1.3 生成top文件等位置限制文件
- 二、定义盒子及添加溶剂
- 2.1 定义盒子
- 2.2 加入溶剂
- 三、添加离子
- 3.1 使用mdp参数文件生成tpr文件
- 3.2 离子的加入
- 3.3 添加离子特别注意事项
- 四、能量最小化
- 4.1 用mdp配置文件生成能量最小化的tpr文件
- 4.2 进行能量最小化处理
- 4.3 可以做个图看一下最小化的过程
- 五、平衡模拟
- 5.1 nvt模拟
- 5.1.1 用mdp配置文件生成nvt的tpr文件
- 5.1.2 进行nvt模拟
- 5.1.3 画图查看nvt平衡过程
- 5.2 npt平衡
- 5.2.1 用mdp配置文件生成npt的tpr文件
- 5.2.2 进行npt模拟
- 5.2.3 画图查看npt平衡过程
- 六、成品模拟
- 6.1 为MD配置mdp文件
- 6.2 开始MD模拟
- 七、分析结果
- 7.1 修正坐标
- 7.2 RMSD分析
- 7.3 Rg分析
- 总结
前言
因为我觉得GROMACS官方英文教程和李继存老师的GROMACS的中文教程对于新手非常不友好,并且有很多细节的地方并没有详细说明,导致有很多困难的地方。这里记录一下自己成功的实验案例。
系统环境
电脑系统:win11(大部分操作) linux:ubuntu22.04 (小部分操作)
我知道gromacs很多在linux上搞,但我就要在windows上搞,windows可以很方便的可视化
python环境:3.6.13
大家还是最好创建一个新的conda环境,我以前就是用的3.11的python结果很多地方报错,最好还是和我用的版本一样
gromacs版本:gmx2020.6_AVX2_CUDA_win64
英文官方教程推荐using a GROMACS version in the 2018.x series,别用老的,老的以前不能使用gpu加速,之后处理会非常慢,除非你用超算,但这就不属于个人电脑能处理的,我是希望能在自己的电脑上跑出来
GPU:GTX1060 6G
相当老的甜品级显卡了,不要用AMD的显卡就行
重要软件:VSCode
我们很多操作都在用VSCode打开并且修改,必须要有,vscode里也装上gromacs的插件,更好看一点
其他软件:pymol3.8 VMD Avogadro(前两个是为了可视化,最后一个这个教程不是必须)
如果环境不会搭的话我会再出一期专门配置环境的,环境配置并不是我们这次教程的重点
这章GROMACS英文教程地址:[GROMACS英文教程地址](http://www.mdtutorials.com/gmx/lysozyme/index.html)
特别强调
我们所有在的命令操作都要在工作目录中进行!
比如我的下载的文件在D:\Code\ChemCode\Gromacs\test001\resource这个文件夹中。
无论你采用那个命令输入方式(cmd或者用powershell或者用anaconda prompt)
都要提前切换到工作目录中,再进行命令的输入
这是我放文件的地方
点击这里输入cmd

这样才算进入工作目录

如果你是win+r然后输入cmd
这并不是我们的工作目录
输入
cd /d D:\Code\ChemCode\Gromacs\test001\resource

来切换到我们的工作目录。
我比较推荐大家使用conda环境中的anaconda prompt,因为后面涉及一些python的操作。打开anaconda prompt,创建你需要的python环境,激活。操作和cmd中一致。我就不放图了。
一、预处理阶段
我先放英文教程,再说我的。

这次处理的是针对egg white lysozyme (PDB code 1AKI),我们要在PCSB PDB这个网站中下载我们需要的1AKI蛋白
搜索1AKI点进去
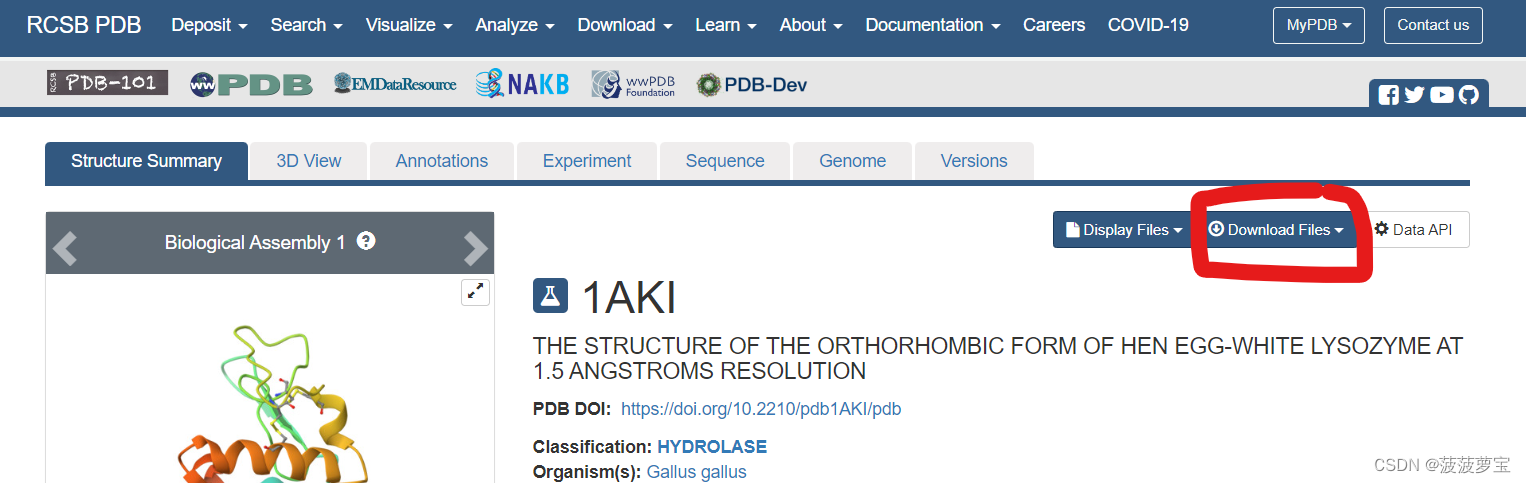
点击downloadfiles
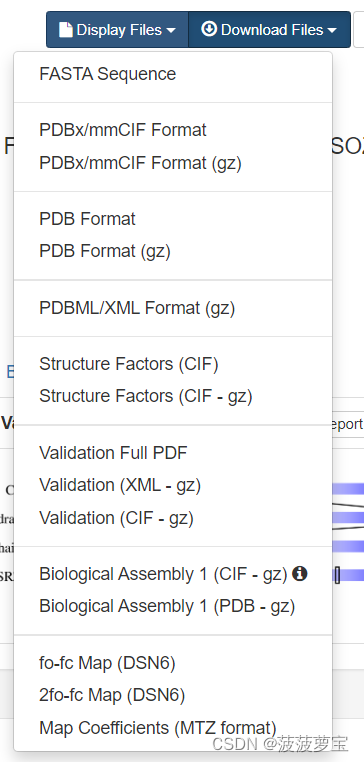
我们下载PDB format格式即可
1AKI这是单链在下载PDB format无所谓 ,后缀有gz是在linux上使用,如果我们要处理的蛋白质是多链的话,若多链下载时注意使用Biological assembly
1.1 补全原子或残基
我们有时候下载的pdb文件中有MISSING字段。用vscode打开下载的pdb,ctrl+f后输入MISSING,并没有MISSING字段。但是在别的pdb文件中可能有,没有的话就跳过这一步。
如果有MISSING字段
使用spdbv打开下载的pdb

这里我用的4NRM蛋白

一打开提示有缺失原子
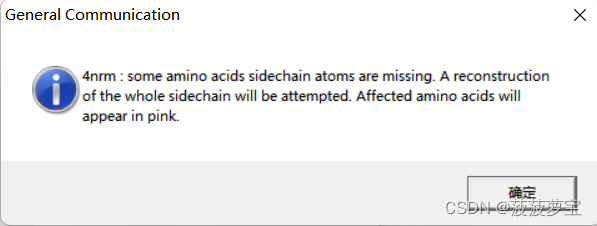
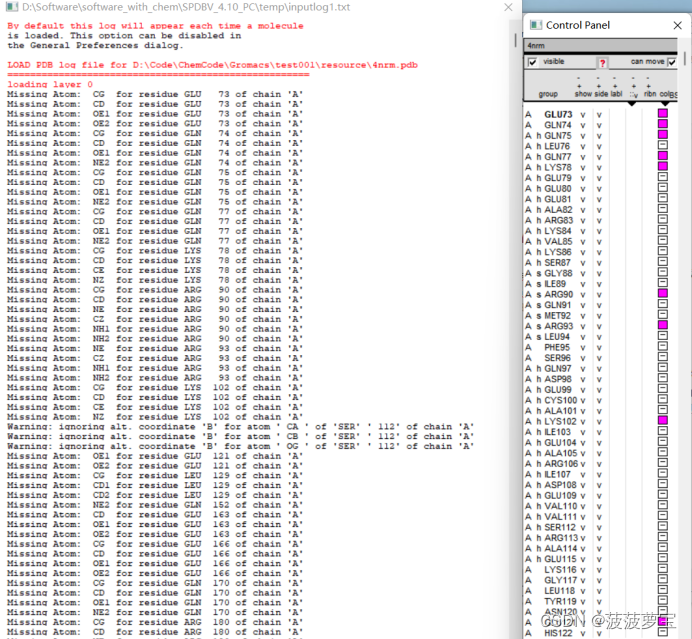
spdbv自动会补全缺失原子,粉色为补全原子
针对缺失残基:1、同源模建 2、直接在C端加上氧原子(在bulid里点击add C-termnal Oxygen)
我推荐针对缺失残基使用第二种方法。

之后点击file然后save as保存为pdb文件即可。之后操作在保存的文件中进行。
1.2 删除水分子
涉及命令
linux:grep -v HOH 1aki.pdb > 1AKI_clean.pdb
如果我们在linux上执行,直接在我们的工作目录中打开终端,然后输入这个命令,我解释一下,grep是搜索,-v是反选,搜索1aki.pdb中除了HOH以外的文本把他们输出成1AKI_clean.pdb这个文件,我们如果用windows中无论是cmd命令窗口还是powershell中都无法识别grep,所以我们要在windows中处理
Windows:需要我们手动处理,TER以下的水分子直接删
用vscode打开我们下载的1aki.pdb文件

我们把他复制一下,重命名为1aki_clean.pdb
重命名是为了不破坏源文件,注意我们之后的所有操作都是名字和命令符要对应上的

打开1aki_clean.pdb后长这样
我这里打开的是1aki.pdb,是为了给你们看一下有HOH的样子,你们操作的时候在1aki_nonHOH.pdb中操作
往下拉,找到终止位TER
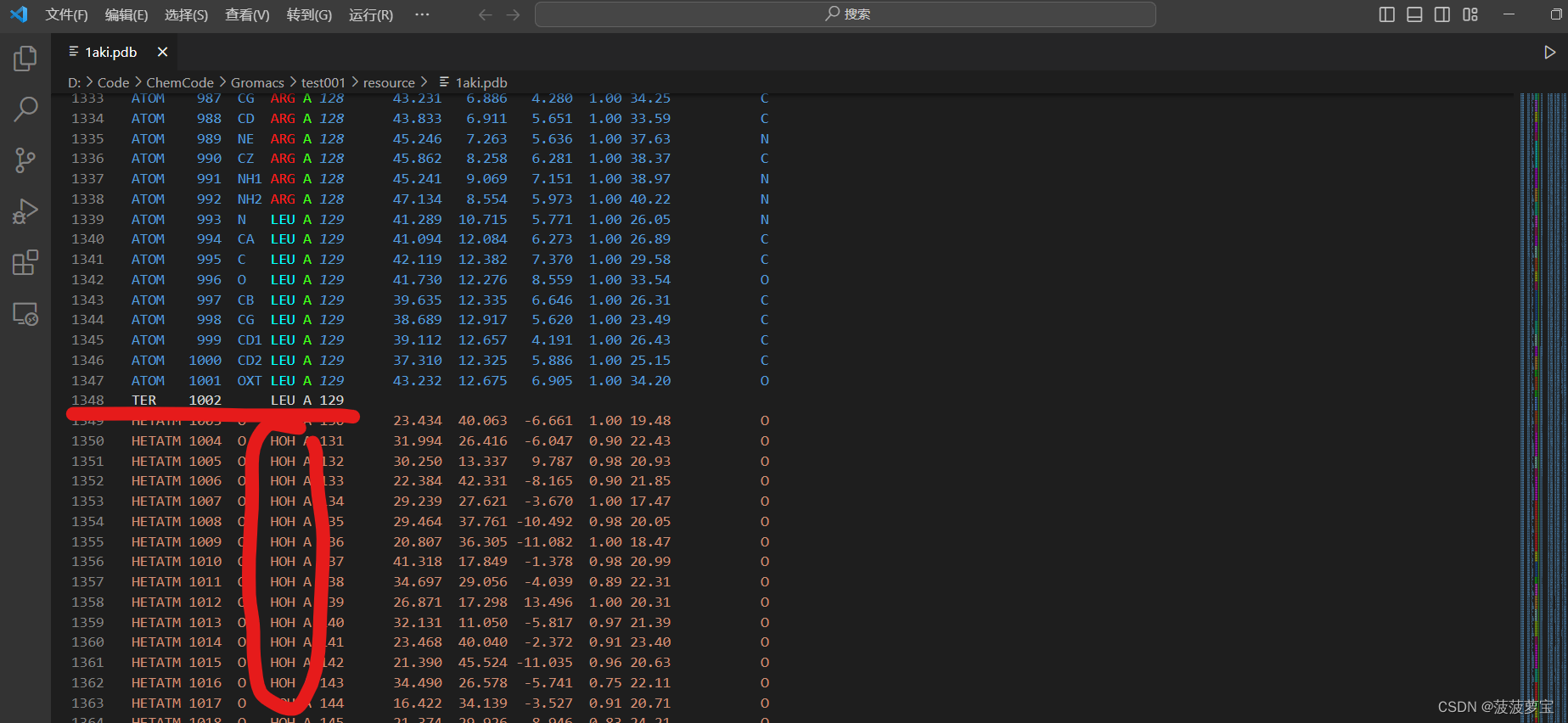
直接删除HOH所在的行,注意是删HOH所在行,别删成别的了,删到CONECT之前就行了,ctrl+s保存即可。
1.3 生成top文件等位置限制文件

gmx pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce

我这里处理完水后的名字叫1AKI_noHOH.pdb,你换成你们处理完水后的pdb文件名就行
顺便解释一下命令的意思,用1AKI_clean.pdb生成gromacs文件1AKI_processed.gro,并选择相应力场及水模型
之后选择力场从而生成拓扑文件
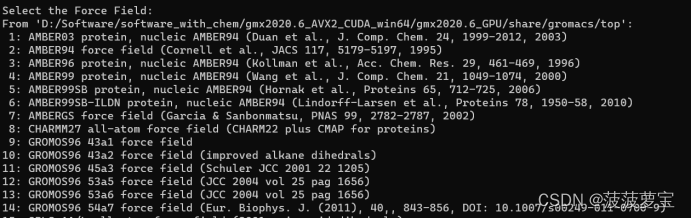
Amber力场比较规范化,若涉及有机小分子优先使用amber力场
使用的amber03的立场和spce的水模型,也就是选择相应的数字输入即可。
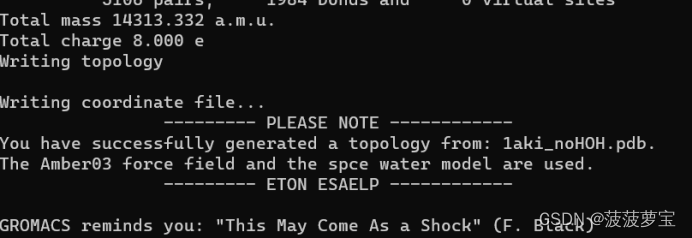
输出成功后显示总电荷为8e 成功生成
输出这三个文件

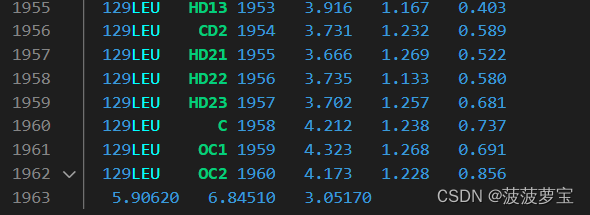
.gro第一个是结构文件,用vscode打开可以看到,是根据残基将其他分类包含残基和坐标信息,最后一行是盒子大小。相比pdb文件只包含原子信息
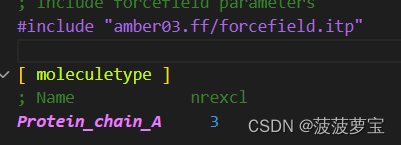
.top代表拓扑文件,记录力场信息和分子类型,原子信息包括原子类型、原子电荷、原子质量等
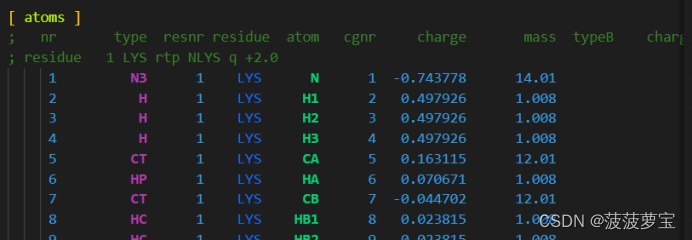
还包括键的参数

.itp位置限制文件,itp是top的一部分,其中还包括水分子的拓扑信息
如果还有别的分子加入,则必须先有对分子的拓扑,再加上第二个分子的位置限制,这个我们在第五个实验,蛋白质与配体配对时,我们需要手动的添加配体信息。
生成这三个文件,我们预处理阶段算是完成。
二、定义盒子及添加溶剂
2.1 定义盒子
跟着官方教程继续走

gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
用我们生成的gro的结构文件,去生成对应的盒子,cubic是立方体,后面我们也会用到十二面体,-d是设置截断半径,需要在计算效率和计算参数找到一个平衡点,一般为蛋白质自身宽度+一半阶段半径
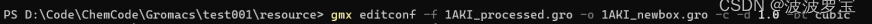
结果
生成这个文件
2.2 加入溶剂

往盒子中添加溶剂
gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top

一共生成生成这些文件
#topol.top.1#是旧的top文件 topol.top是新的top文件
用新生成的topol.top对比我们1.3步之后生成的topol.top文件
相比之前的top多了溶剂分子 SOL就是溶剂水
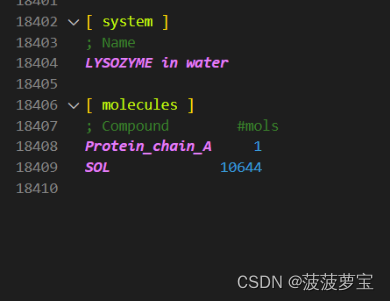
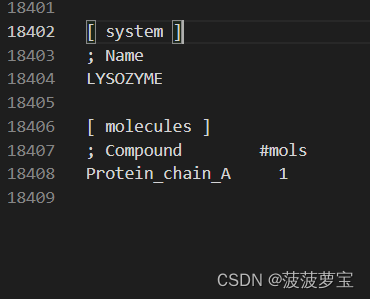
官方教程中有10832个溶剂,而我只有10644个,这个不碍事,可能是我们使用的gromacs2020和2018之间版本的问题
禁止手动修改SOL的数字,否则后续会报错!!!
我们可以用pymol或者vmd来查看生成的盒子信息
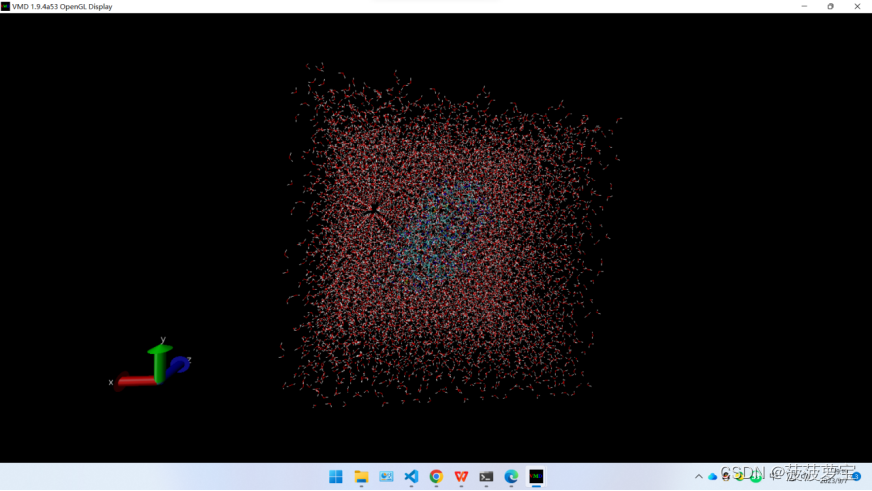
可以看出,蛋白质在盒子最中心,溶剂分子包裹蛋白质
三、添加离子
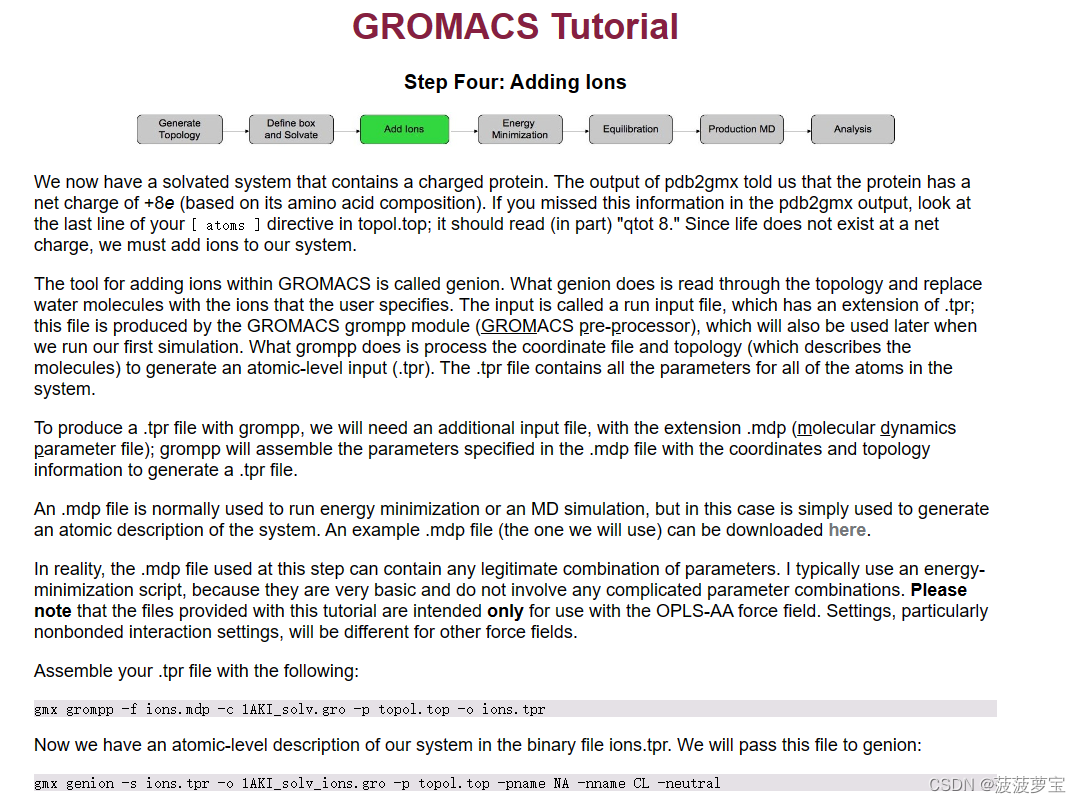
3.1 使用mdp参数文件生成tpr文件
我们在1.3步中得到我们的蛋白质带8e的电荷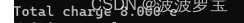
因为现在是一个带电的蛋白质的溶剂体系,由于生命体系中净电荷为0,我们必须将抗衡离子添加到我们的体系中
添加离子的工具称为genion,genion将指定离子替换为水分子并生成tpr文件,采用GROMACS grompp 模块,genion默认使用的盐为NaCl
tpr文件中包含系统中所有原子的参数,若要生成tpr文件,需要mdp文件,grompp 会将 .mdp 文件中指定的参数与坐标和拓扑信息组合在一起,以生成 .tpr 文件
mdp是整个gromacs中最重要需要手动修改的参数文件,需要做出个性化的调整
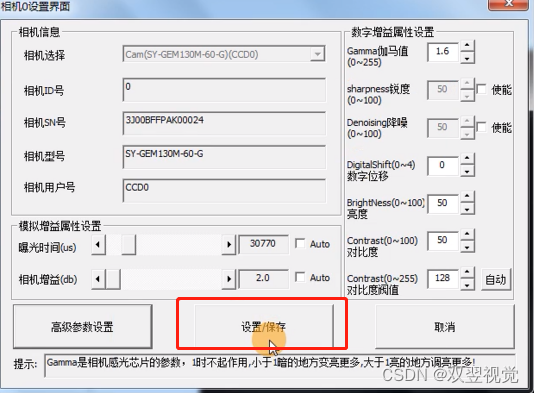
现在我们工作目录中创建一个新文件,命名为ions.mdp。
(我推荐在vscode里打开工作目录,之后的操作就在工作目录中了。)
在工作目录中创建新文件ions.mdp
点击这里,将里面的东西全选(ctrl+a)复制粘贴到ions.mdp中
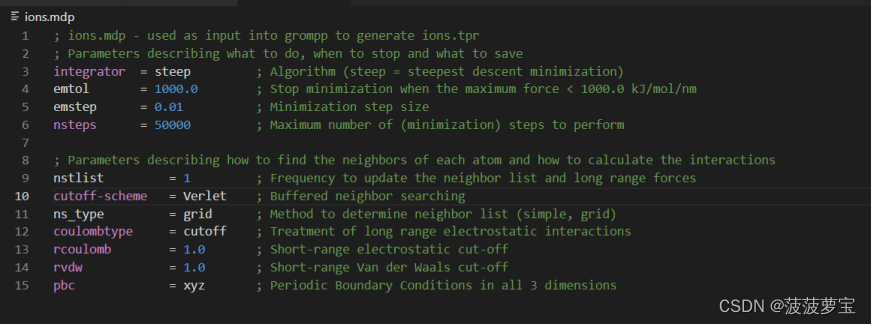
将gro文件与mdp组合在一起生成tpr文件
gmx grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr

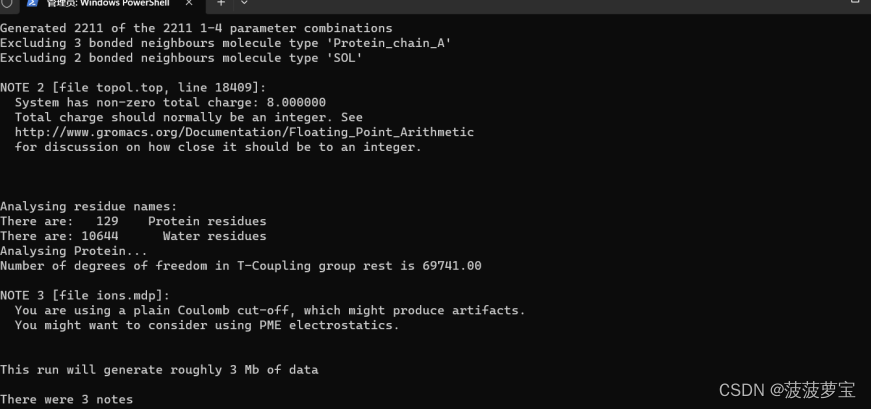
3.2 离子的加入
对二进制文件 ions.tpr 中对体系进行了原子水平的描述,并传递给genion,并且添加pname阳离子NA和nname阴离子CL
gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral

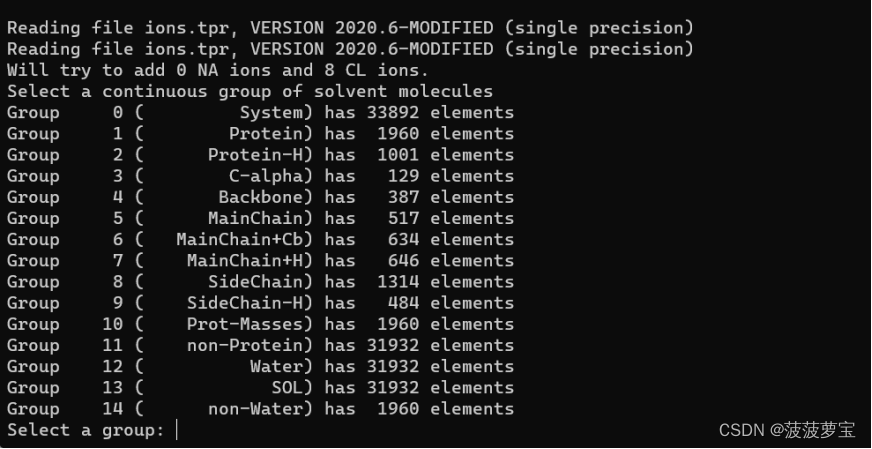
选择13组SOL进行包埋离子,离子要替换掉的对象一定是溶剂
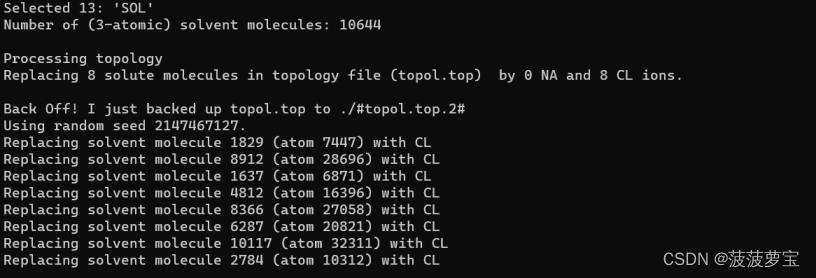
在vscode中查看输出的top文件
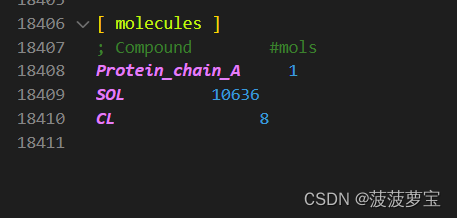
加了8个CL离子来平衡八个正电荷
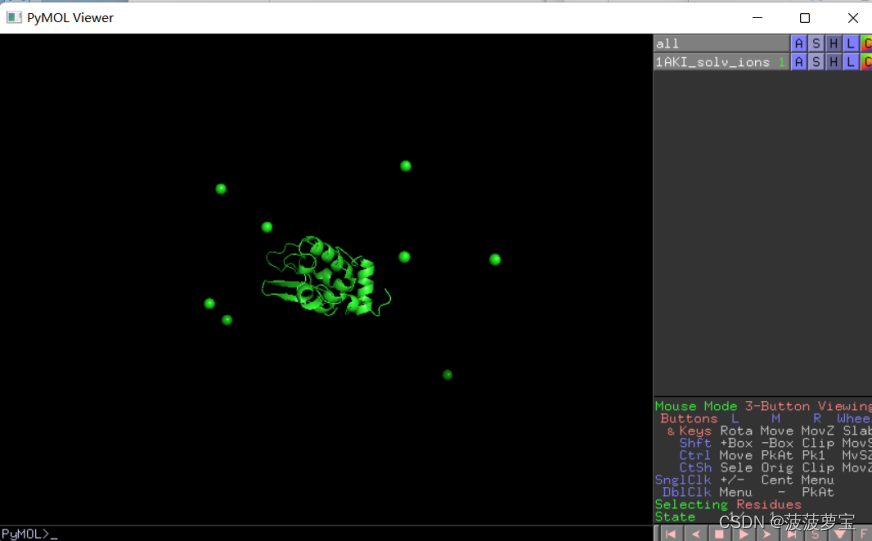
我们用pymol可视化一下,隐藏掉水分子,我们能明显看到有八个CL离子
3.3 添加离子特别注意事项
针对3.2步特别注意,在不同的立场中,相同离子的名称可能不一样,在amber03力场中钠离子就叫NA,氯离子叫CL,但是在有些立场中,例如charmm36立场中,钠离子依然叫NA,但氯离子叫CLA,查看力场在我们gromacs文件夹下,例如我的gromacs2020.6_GPU中,力场文件就在gmx2020.6_GPU\share\gromacs\top这个文件夹下。
因为我们在1.2步选择时amber03力场,打开amber03.ff
其中ions.itp就是保存着对应离子的名称
我们用vscode打开ions.itp
把他拖到vscode图标里就能打开了
ctrl+f搜索NA就能搜到,在amber03力场中氯离子叫CL
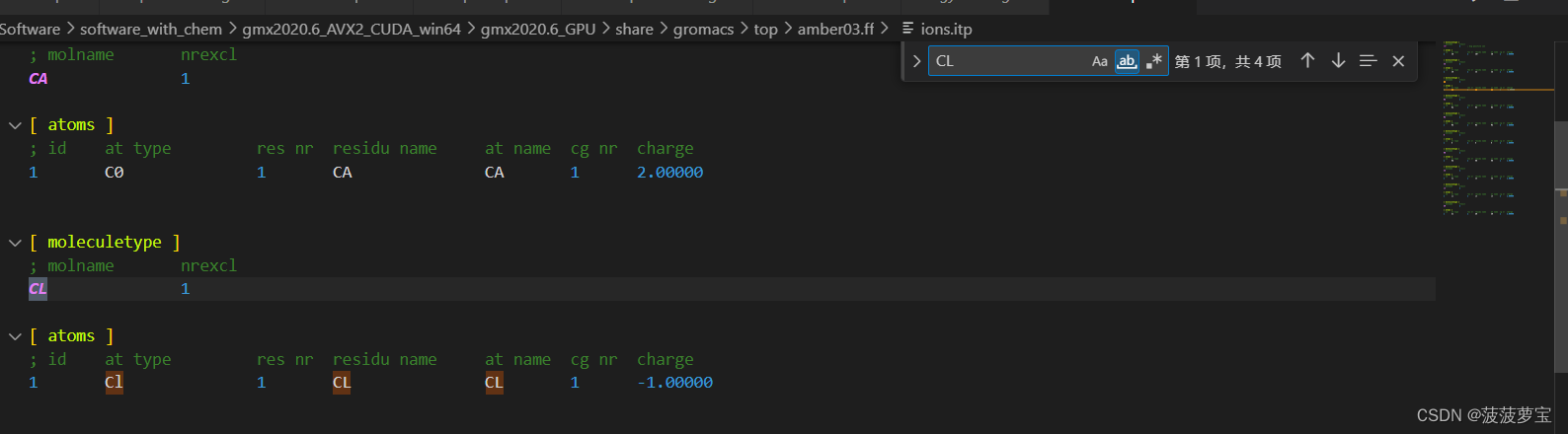
我们用相同的方式打开charmm36力场
这里的氯离子就叫CLA
at type才是计算机识别的名称类型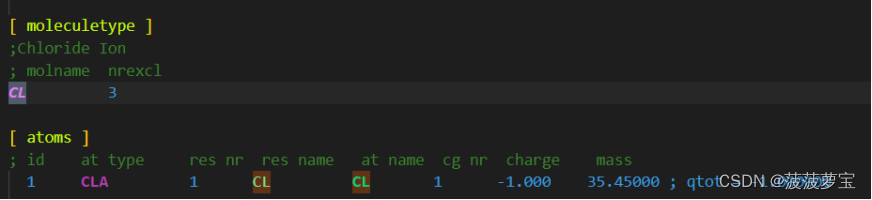
因此我们在添加离子时,要注意力场文件中的原子名称是否对应上,否则gromacs就会报错!
四、能量最小化
4.1 用mdp配置文件生成能量最小化的tpr文件
能量最小化能够有效避免直接模拟后体系崩溃
还是现在工作目录中创建minim.mdp文件
点击这里将里面的东西全选(ctrl+a)复制粘贴到minim.mdp中
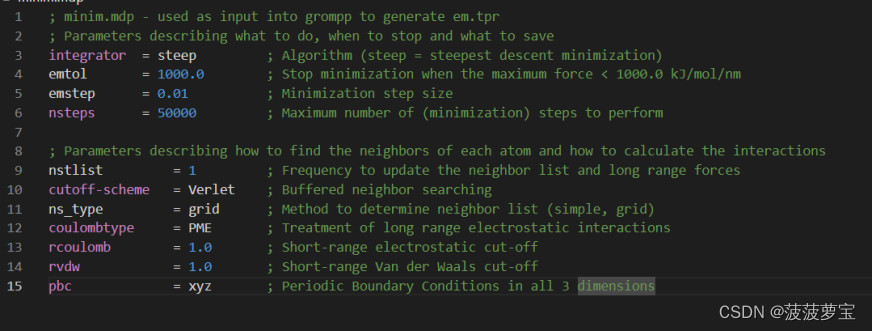
为能量最小化生成tpr文件中包含系统中所有原子的参数
gmx grompp -f minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
生成
4.2 进行能量最小化处理
gmx mdrun -v -deffnm em

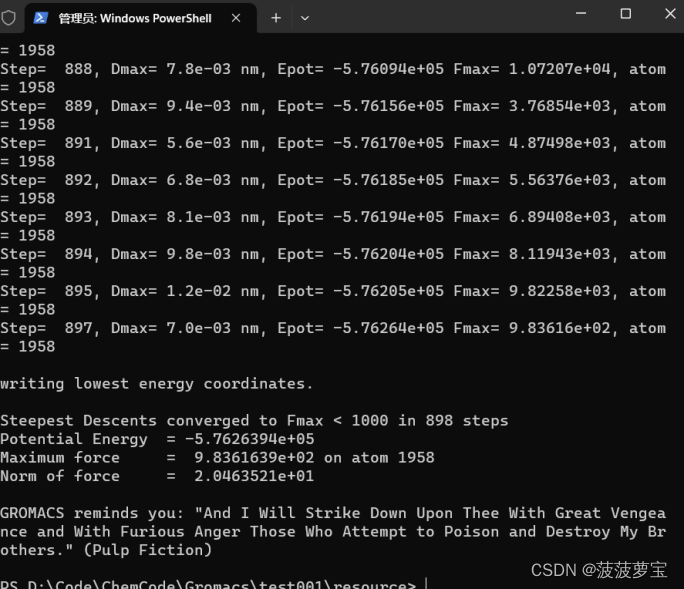 用了897步使得能量小于1000
用了897步使得能量小于1000
生成这些文件

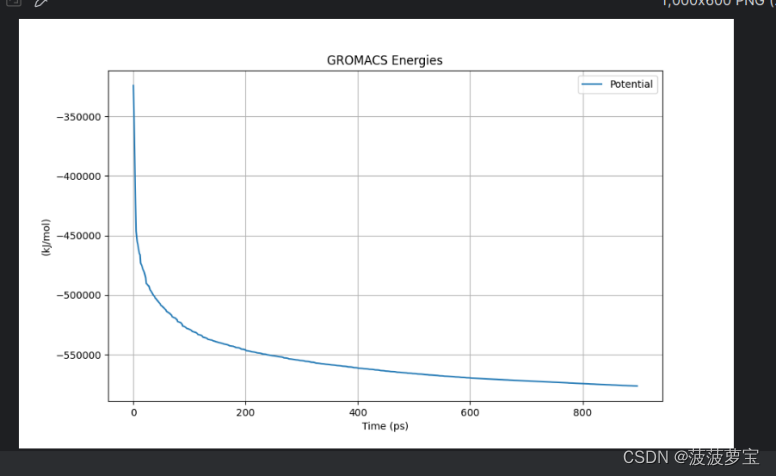
4.3 可以做个图看一下最小化的过程
gmx energy -f em.edr -o potential.xvg
选择10 0
输入10 0 中间有个空格
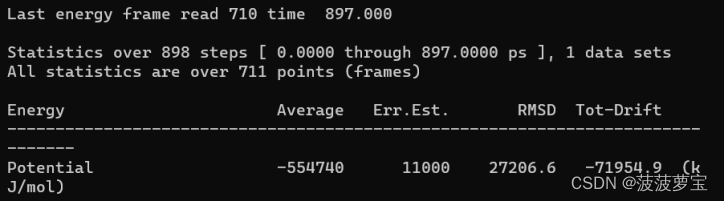
用vscode查看生成的potential.xvg文件
我用python做了图
注释的地方是有的地方用了n步平均值,这里并没有用到我就注释了
后面所有的图都是那这个改的,改改路径什么的大家自己来咯,要改的地方也就FILE_PATH、START_LINE、plt.plot里label的名称、plt.xlabel、plt.ylabel、plt.title、plt.savefig的名称,大家按照xvg的里面的内容自己改一改^_^
import matplotlib.pyplot as plt
import numpy as np
FILE_PATH = r'D:\Code\ChemCode\Gromacs\test001\resource\potential.xvg'
START_LINE = r'@ s0 legend "Potential"'
plt.rcParams['font.sans-serif'] = 'SimHei'
with open(FILE_PATH, 'r') as file:
lines = file.readlines()
x_values = []
y_values = []
data_started = False
for line in lines:
if data_started:
parts = line.split()
if len(parts) >= 2:
x_values.append(float(parts[0]))
y_values.append(float(parts[1]))
elif line.startswith(START_LINE):
data_started = True
plt.figure(figsize=(10, 6))
# plt.xlim(0, max(x_values))
# plt.xticks(np.arange(0, max(x_values)+1, step=20))
plt.plot(x_values, y_values, label='Potential')
plt.xlabel('Time (ps)')
plt.ylabel('(kJ/mol)')
plt.title('GROMACS Energies')
# window_size = 10
# running_average = np.convolve(y_values, np.ones(window_size)/window_size, mode='valid')
# plt.plot(x_values[window_size-1:], running_average, label=f'Running Average ({window_size}-ps window)', color='red')
plt.legend()
plt.grid(True)
plt.savefig('Potential.png', dpi=300, bbox_inches='tight')
plt.show()
看一下做的图
五、平衡模拟

对整个体系进行位置限制性模拟,也就是对溶剂和离子进行弛豫同时保持蛋白质原子的位置不变. 在位置限制性模拟中会限制(或部分冻结)大分子中的原子位置,设置体系的温度和压强,nvt中设置温度,npt中设置压强
5.1 nvt模拟
5.1.1 用mdp配置文件生成nvt的tpr文件
还是现在工作目录中创建nvt.mdp文件
点击这里将里面的东西全选(ctrl+a)复制粘贴到nvt.mdp中
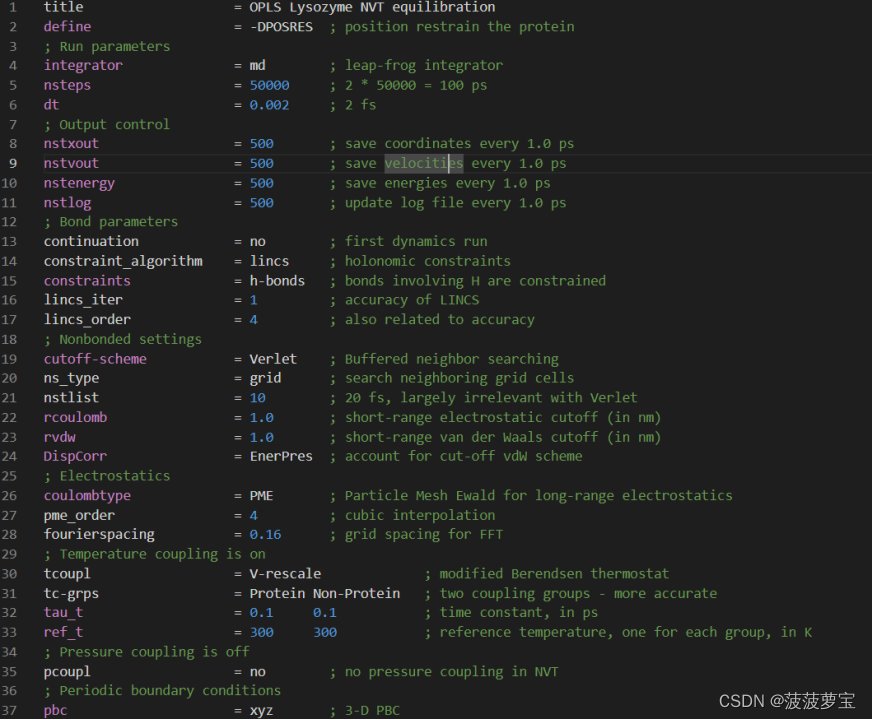
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr

生成
5.1.2 进行nvt模拟
gmx mdrun -v -deffnm nvt

生成
5.1.3 画图查看nvt平衡过程
gmx energy -f nvt.edr -o temperature.xvg

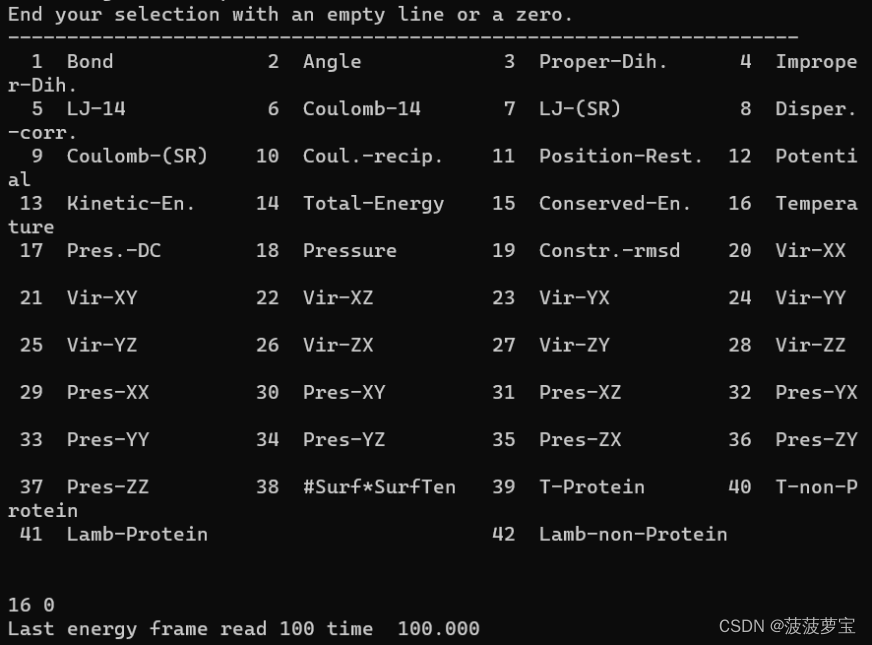
选择16 0
生成

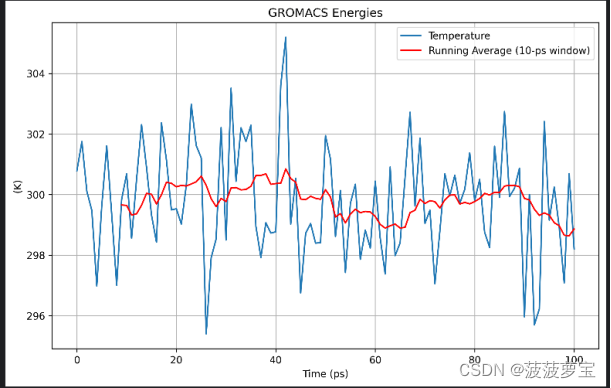
画图xvg
画图的代码大家按照4.3步和新生成的xvg图像自己改一下吧^_^
这里用到了10ps的平均值,大家要把注释掉的部分加回来
5.2 npt平衡
5.2.1 用mdp配置文件生成npt的tpr文件
还是现在工作目录中创建npt.mdp文件
点击这里将里面的东西全选(ctrl+a)复制粘贴到npt.mdp中
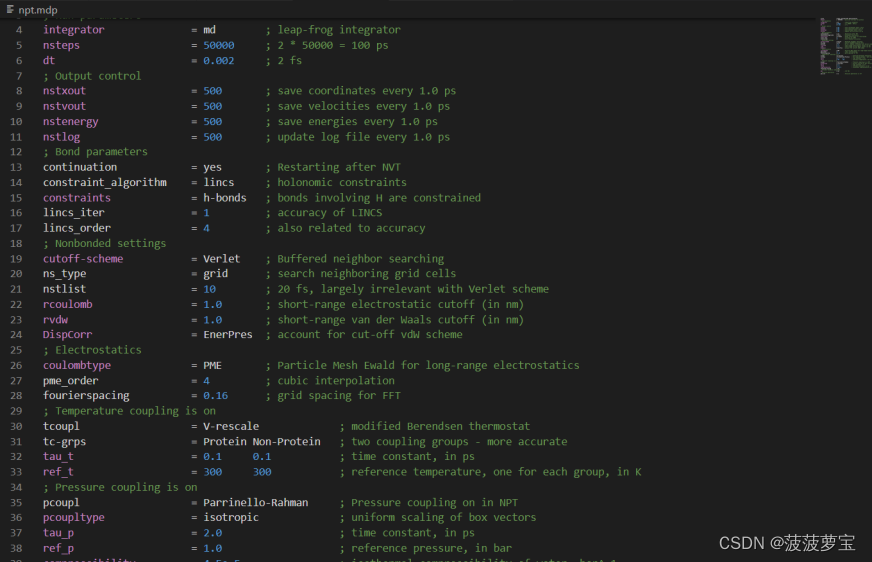
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr

生成
5.2.2 进行npt模拟
gmx mdrun -v -deffnm npt

生成
5.2.3 画图查看npt平衡过程
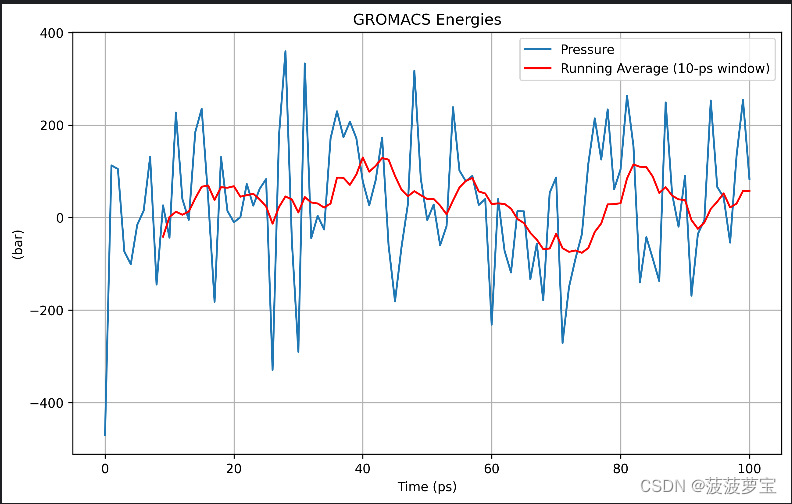
gmx energy -f npt.edr -o pressure.xvg

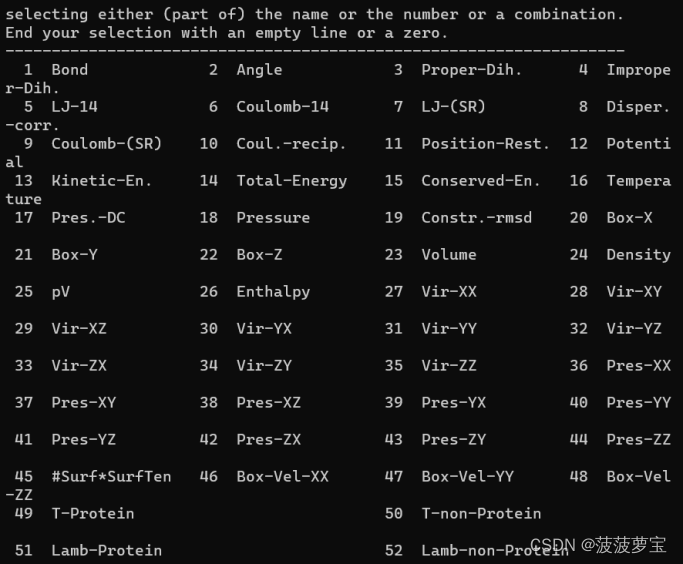
选18 0
生成
画图xvg
画图的代码大家按照4.3步和新生成的xvg图像自己改一下吧^_^
这里用到了10ps的平均值,大家要把注释掉的部分加回来
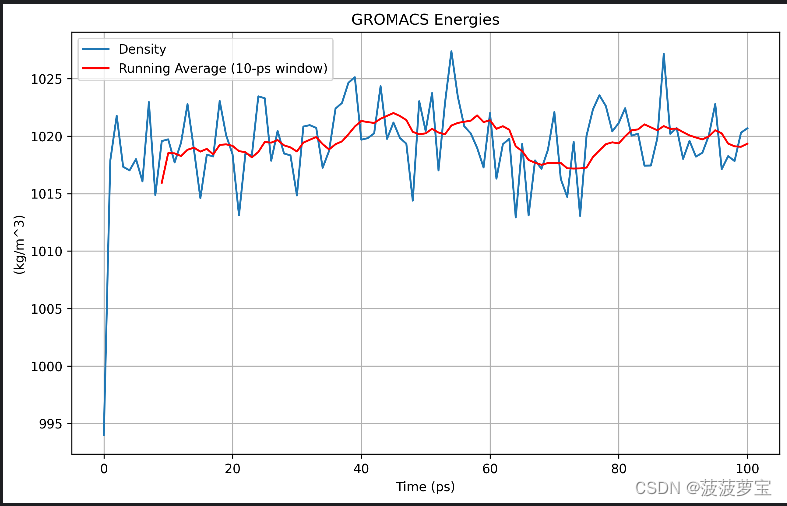
还有个density的图
gmx energy -f npt.edr -o density.xvg

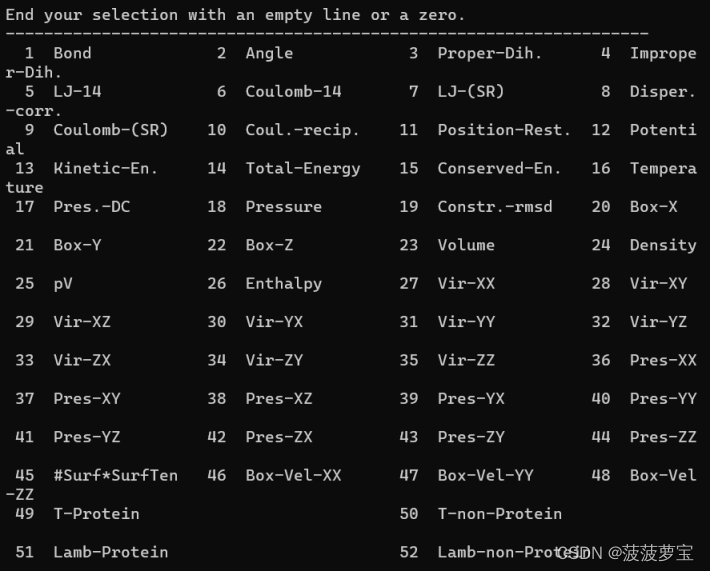
选24 0
画图
六、成品模拟
6.1 为MD配置mdp文件
还是现在工作目录中创建md.mdp文件
点击这里将里面的东西全选(ctrl+a)复制粘贴到md.mdp中
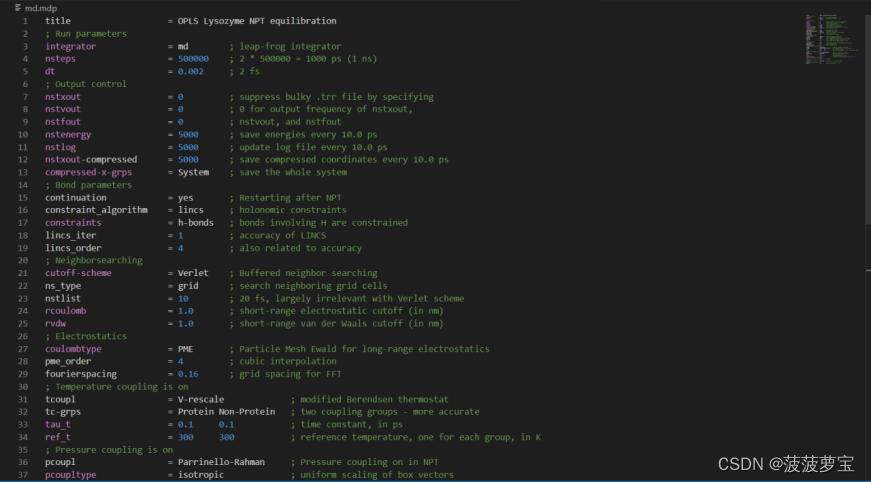
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr

生成
6.2 开始MD模拟
我们的初衷就是在gpu上模拟,所以在后面选项加上-nb gpu
gmx mdrun -v -deffnm md_0_1 -nb gpu
因为我们md.mdp文件中nsteps设置的50w,50w steps在我的1060上预计10分钟跑完
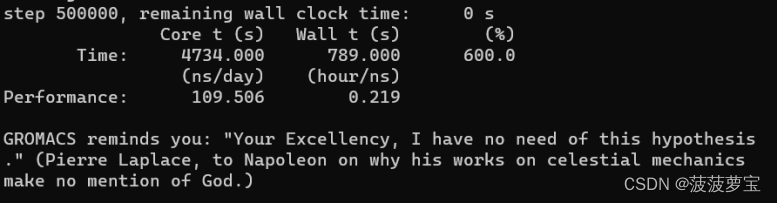
生成
七、分析结果
RMSD 均方根偏差:反应某时刻结构相对于初始构象的偏差
RMSF 氨基酸残基的波动:每个氨基酸残基随时间的平均偏移参考位置的情况
RMSF值高说明该氨基酸或基团表现出较大的柔韧性,反之则柔韧性较差
7.1 修正坐标
因为我们在创建盒子时,有些原子处于盒子的边缘,一半在盒子内,一半在盒子外(也不是在盒子外,在另一个盒子里探出头,这个涉及到PBC周期型边界条件,有机会再讲)
使用trjconv来手动提取坐标、纠正周期性或者手动调整轨迹,使用trjconv消除体系的周期性
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -center

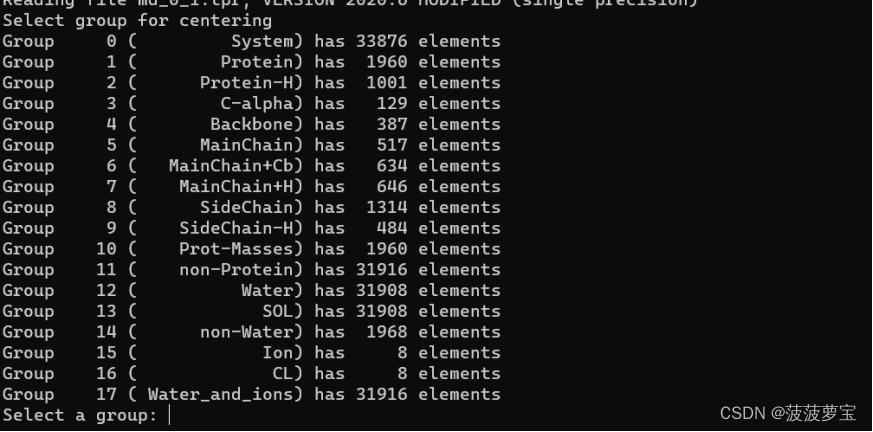
选择1作为体系进行偏移
选择0为输出结果

矫正之后的模拟结果

7.2 RMSD分析
获取RMSD的xvg的文件
gmx rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg -tu ns

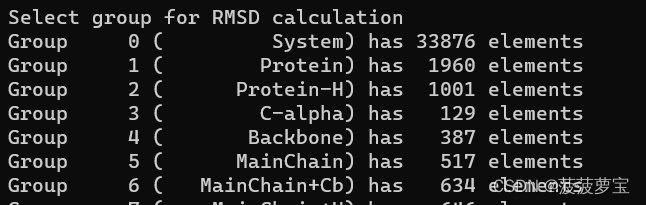
选择4 backbone作为最小二乘法拟合以及RMSD计算
获得RMSD的xvg的文件

也可以获取相对于结晶结构的RMSD
gmx rms -s em.tpr -f md_0_1_noPBC.xtc -o rmsd_xtal.xvg -tu ns
画图
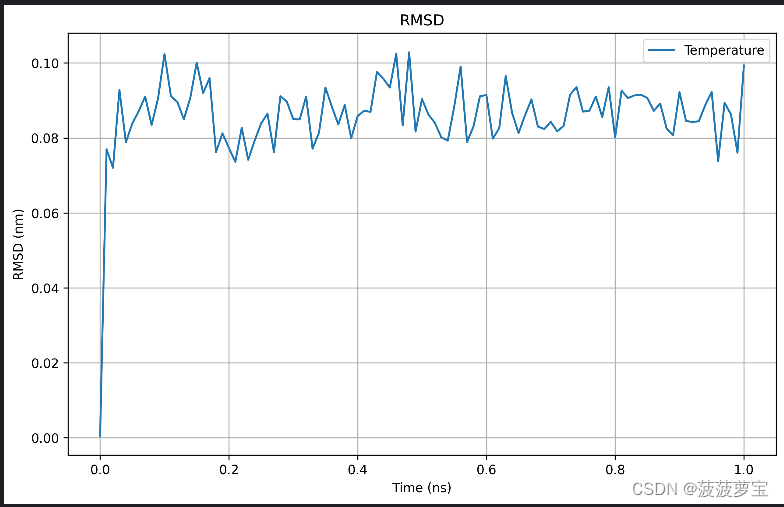
基本在0.1nm 1A以下,相当稳定
7.3 Rg分析
查看蛋白质的均方回转半径(Rg)可以描述其紧密程度,如果蛋白质处于稳固的折叠状态,Rg会处于一个相对稳定值。如果蛋白没有折叠,Rg会一直变化。
gmx gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o gyrate.xvg
选择1 针对蛋白质
生成
画图
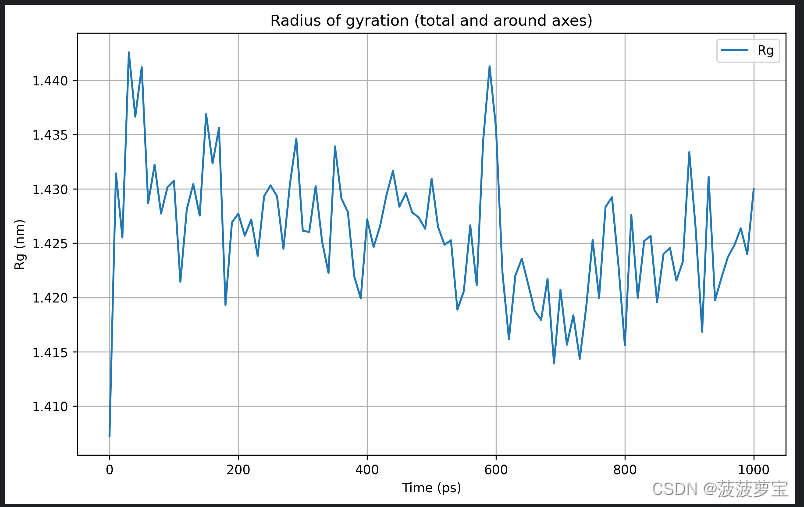
从Rg相对合理的稳定值可以发现,在300K温度下1ns的模拟中,蛋白质处于非常稳定的折叠状态。
总结
到这里第一章关于egg white lysozyme的gromacs教程就结束啦,因为我的专业是软件工程,gromacs有时候科研的时候会用到,踩了很多坑,希望学弟学妹们就别绕远路了。关于第五章和环境配置的部分我有时间再写,如果反响好的话。